 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3gsb: CRYSTAL STRUCTURE OF GLUTAMATE-1-SEMIALDEHYDE AMINOMUTASE IN COMP... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3gsb | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF GLUTAMATE-1-SEMIALDEHYDE AMINOMUTASE IN COMPLEX WITH GABACULINE | |||||||||
 Components Components | PROTEIN (GLUTAMATE SEMIALDEHYDE AMINOTRANSFERASE) | |||||||||
 Keywords Keywords | ISOMERASE / CHLOROPHYLL BIOSYNTHESIS / PYRIDOXAL-5'-PHOSPHATE / PYRIDOXAMINE-5'-PHOSPHATE / ASYMMETRIC DIMER / GABACULINE | |||||||||
| Function / homology |  Function and homology information Function and homology informationglutamate-1-semialdehyde 2,1-aminomutase / glutamate-1-semialdehyde 2,1-aminomutase activity / chlorophyll biosynthetic process / : / transaminase activity / pyridoxal phosphate binding / cytoplasm Similarity search - Function | |||||||||
| Biological species |  Synechococcus sp. (bacteria) Synechococcus sp. (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / Resolution: 3 Å X-RAY DIFFRACTION / Resolution: 3 Å | |||||||||
 Authors Authors | Hennig, M. / Jansonius, J.N. | |||||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 1997 Title: Crystal structure of glutamate-1-semialdehyde aminomutase: an alpha2-dimeric vitamin B6-dependent enzyme with asymmetry in structure and active site reactivity. Authors: Hennig, M. / Grimm, B. / Contestabile, R. / John, R.A. / Jansonius, J.N. #1: Journal: J.Mol.Biol. / Year: 1994Title: Crystallization and Preliminary X-Ray Analysis of Wild-Type and K272A Mutant Glutamate 1-Semialdehyde Aminotransferase from Synechococcus Authors: Hennig, M. / Grimm, B. / Jenny, M. / Muller, R. / Jansonius, J.N. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3gsb.cif.gz | 173.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3gsb.ent.gz | 137.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3gsb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gs/3gsbftp://data.pdbj.org/pub/pdb/validation_reports/gs/3gsb | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | THE ASYMMETRIC UNIT CONTAINS A STRUCTURALLY ASYMMETRIC HOMODIMER. |
-Components
| #1: Protein | Mass: 46059.523 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Synechococcus sp. (bacteria) / Strain: GR6 / Production host: References: UniProt: P24630, glutamate-1-semialdehyde 2,1-aminomutase #2: Chemical | 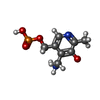  Mass: 248.173 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H13N2O5P Mass: 248.173 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H13N2O5P#3: Chemical | ChemComp-GAB / | 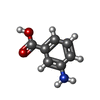  Mass: 137.136 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H7NO2 Mass: 137.136 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H7NO2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 50 % Description: COFACTORS AND WATERS WERE REMOVED FROM THE MODEL | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 Details: 50MM NA-CACODYLATE BUFFER PH 7.0, 200 MM MG-ACETATE, 19.5% PEG 10,000, SOAKING CONDITIONS: 3MM GABACULINE FOR 4 DAYS | |||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, sitting drop / Details: Hennig, M., (1994) J.Mol.Biol., 242, 591. | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 273 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ELLIOTT GX-21 / Wavelength: 1.54 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 18, 1995 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 3→15 Å / Num. obs: 18662 / % possible obs: 98 % / Redundancy: 3.1 % / Biso Wilson estimate: 32 Å2 / Rsym value: 0.114 / Net I/σ(I): 6.2 |
| Reflection shell | Resolution: 3→3.16 Å / Redundancy: 3.1 % / Mean I/σ(I) obs: 1.8 / Rsym value: 0.412 / % possible all: 96.4 |
| Reflection | *PLUS Num. measured all: 57653 / Rmerge(I) obs: 0.114 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Starting model: GSAT WILD-TYPE (NATIVE) STRUCTURE Resolution: 3→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3→3.05 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 5 % / Rfactor Rfree: 0.25 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 33.2 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.38 / % reflection Rfree: 5 % / Rfactor Rwork: 0.28 |
