 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3es0: A bimolecular anti-parallel-stranded Oxytricha nova telomeric qua... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3es0 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | A bimolecular anti-parallel-stranded Oxytricha nova telomeric quadruplex in complex with a 3,6-disubstituted acridine BSU-6048 | ||||||||||||||||||
 Components Components | 5'-D(* Keywords KeywordsDNA / quadruplex / Oxytricha nova / BSU-6048 / BSU6048 / anti-parallel / bimolecular / macromolecule / G-quadruplex | Function / homology | : / Chem-NCK / DNA / DNA (> 10) |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å  Authors AuthorsCampbell, N.H. / Parkinson, G. / Neidle, S. |  CitationJournal: Biochemistry / Year: 2009 CitationJournal: Biochemistry / Year: 2009Title: Selectivity in Ligand Recognition of G-Quadruplex Loops. Authors: Campbell, N.H. / Patel, M. / Tofa, A.B. / Ghosh, R. / Parkinson, G.N. / Neidle, S. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3es0.cif.gz | 25.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3es0.ent.gz | 18 KB | Display | PDB format |
| PDBx/mmJSON format | 3es0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/es/3es0ftp://data.pdbj.org/pub/pdb/validation_reports/es/3es0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3em2C  3eqwC  3eruC  3et8C  3euiC  3eumC  1l1hS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |





-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 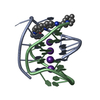
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 3805.460 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: The sequence occurs naturally in Oxytricha nova #2: Chemical | ChemComp-K /   Mass: 39.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: K#3: Chemical | ChemComp-NCK / |   Mass: 515.690 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H41N5O2 Mass: 515.690 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H41N5O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 56 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 56 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.11 Å3/Da / Density % sol: 41.82 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 285.15 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 2 microliter drops containing 5% v/v MPD, 0.50 mM DNA, 0.25 mM Ligand, 40 mM Potassium chloride, 5 mM Magnesium chloride, 4.1 Spermine equilibrated against 35% v/v MPD, pH 7.0, VAPOR ...Details: 2 microliter drops containing 5% v/v MPD, 0.50 mM DNA, 0.25 mM Ligand, 40 mM Potassium chloride, 5 mM Magnesium chloride, 4.1 Spermine equilibrated against 35% v/v MPD, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 285.15K | ||||||||||||||||||||||||
| Components of the solutions |
|
-Data collection
| Diffraction | Mean temperature: 105 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Nov 2, 2006 / Details: mirrors |
| Radiation | Monochromator: Osmic mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→24.5 Å / Num. all: 3561 / Num. obs: 3549 / % possible obs: 99.7 % / Observed criterion σ(I): 3 / Redundancy: 2.91 % / Biso Wilson estimate: 27.135 Å2 / Rmerge(I) obs: 0.041 / Net I/σ(I): 18.1 |
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 2.93 % / Rmerge(I) obs: 0.085 / Mean I/σ(I) obs: 10.2 / Num. unique all: 348 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1L1H Resolution: 2.2→24.5 Å / Cor.coef. Fo:Fc: 0.942 / Cor.coef. Fo:Fc free: 0.895 / SU B: 5.344 / SU ML: 0.134 / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / ESU R: 0.344 / ESU R Free: 0.232 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 11.105 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→24.5 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.257 Å / Total num. of bins used: 20
|
