 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1oce | ||||||
|---|---|---|---|---|---|---|---|
| Title | ACETYLCHOLINESTERASE (E.C. 3.1.1.7) COMPLEXED WITH MF268 | ||||||
 Components Components | ACETYLCHOLINESTERASE | ||||||
 Keywords Keywords | HYDROLASE / CARBOXYLIC ESTERASE / NEUROTRANSMITTER CLEAVAGE / SERINE ESTERASE | ||||||
| Function / homology |  Function and homology information Function and homology informationacetylcholine catabolic process in synaptic cleft / acetylcholinesterase / choline metabolic process / acetylcholinesterase activity / side of membrane / synaptic cleft / synapse / : / plasma membrane Similarity search - Function | ||||||
| Biological species |   Torpedo californica (Pacific electric ray) Torpedo californica (Pacific electric ray) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å | ||||||
 Authors Authors | Bartolucci, C. / Perola, E. / Cellai, L. / Brufani, M. / Lamba, D. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1999 Title: "Back door" opening implied by the crystal structure of a carbamoylated acetylcholinesterase. Authors: Bartolucci, C. / Perola, E. / Cellai, L. / Brufani, M. / Lamba, D. #1: Journal: Biochim.Biophys.Acta / Year: 1997Title: Long Chain Analogs of Physostigmine as Potential Drugs for Alzheimer'S Disease: New Insights Into the Mechanism of Action in the Inhibition of Acetylcholinesterase Authors: Perola, E. / Cellai, L. / Lamba, D. / Filocamo, L. / Brufani, M. #2: Journal: J.Neurosci.Res. / Year: 1996Title: Effects of Mf-268, a New Cholinesterase Inhibitor, on Acetylcholine and Biogenic Amines in Rat Cortex Authors: Zhu, X.D. / Cuadra, G. / Brufani, M. / Maggi, T. / Pagella, P.G. / Williams, E. / Giacobini, E. #3: Journal: Bioorg.Med.Chem.Lett. / Year: 1995Title: Synthesis and Structure-Activity Relationships of New Acetylcholinesterase Inhibitors: Morpholinoalkylcarbamoyloxyeseroline Derivatives Authors: Alisi, M.A. / Brufani, M. / Filocamo, L. / Gostoli, G. / Licandro, E. / Cesta, M.C. / Lappa, S. / Marchesini, D. / Pagella, P. #4: Journal: Science / Year: 1991Title: Atomic Structure of Acetylcholinesterase from Torpedo Californica: A Prototypic Acetylcholine-Binding Protein Authors: Sussman, J.L. / Harel, M. / Frolow, F. / Oefner, C. / Goldman, A. / Toker, L. / Silman, I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1oce.cif.gz | 121 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1oce.ent.gz | 92.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1oce.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/oc/1oceftp://data.pdbj.org/pub/pdb/validation_reports/oc/1oce | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2aceS S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 60736.516 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: INTER-MONOMER DISULFIDE BRIDGE Source: (natural) Torpedo californica (Pacific electric ray)Organ: ELECTRIC ORGAN / Variant: G2 FORM / Tissue: ELECTROPLAQUE / References: UniProt: P04058, acetylcholinesterase |
|---|---|
| #2: Chemical | ChemComp-MF2 / 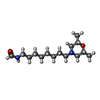  Mass: 270.411 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H30N2O2 Mass: 270.411 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H30N2O2 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.98 Å3/Da / Density % sol: 68.8 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6 / Details: pH 6.0 | |||||||||||||||||||||||||
| Crystal | *PLUS | |||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop / Details: Sussman, J.L., (1988) J. Mol. Biol., 203, 821. | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ELETTRA  / Beamline: 5.2R / Wavelength: 0.92 / Beamline: 5.2R / Wavelength: 0.92 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE AREA DETECTOR / Date: Jun 1, 1997 / Details: TOROIDAL MIRROR |
| Radiation | Monochromator: SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.92 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→20 Å / Num. obs: 24049 / % possible obs: 87.2 % / Observed criterion σ(I): 0 / Redundancy: 2.3 % / Biso Wilson estimate: 42.2 Å2 / Rsym value: 0.14 / Net I/σ(I): 6.7 |
| Reflection shell | Resolution: 2.7→2.8 Å / Redundancy: 1.6 % / Mean I/σ(I) obs: 1.7 / Rsym value: 0.648 / % possible all: 69.7 |
| Reflection | *PLUS Num. measured all: 248837 / Rmerge(I) obs: 0.14 |
| Reflection shell | *PLUS % possible obs: 69.7 % / Num. unique obs: 1886 / Rmerge(I) obs: 0.648 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2ACE Resolution: 2.7→8 Å / Rfactor Rfree error: 0.009 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.3 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.7→2.86 Å / Rfactor Rfree error: 0.034 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
