 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1me4: High Resolution Crystal Structure Analysis Of Cruzain non-covalen... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1me4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | High Resolution Crystal Structure Analysis Of Cruzain non-covalently Bound To A Hydroxymethyl Ketone Inhibitor (I) | ||||||
 Components Components | CRUZIPAIN | ||||||
 Keywords Keywords | HYDROLASE / cysteine protease / non-covalent inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.2 Å | ||||||
 Authors Authors | Brinen, L.S. / Huang, L. / Ellman, J.A. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem. / Year: 2003 Title: Crystal Structures of Reversible Ketone-based Inhibitors of the Cysteine Protease Cruzain Authors: Huang, L. / Brinen, L.S. / Ellman, J.A. #1: Journal: Structure / Year: 2000Title: A target within the target: probing cruzain's P1' site to define structural determinants for the Chagas' disease protease. Authors: Brinen, L.S. / Hansell, E. / Cheng, J. / Roush, W.R. / McKerrow, J.H. / Fletterick, R.J. #2: Journal: J.Mol.Biol. / Year: 1995Title: THE CRYSTAL STRUCTURE OF CRUZAIN: A THERAPEUTIC TARGET FOR CHAGAS' DISEASE Authors: MCGRATH, M.E. / EAKIN, A.E. / ENGEL, J.C. / MCKERROW, J.H. / CRAIK, C.S. / FLETTERICK, R.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1me4.cif.gz | 116.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1me4.ent.gz | 88.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1me4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/me/1me4ftp://data.pdbj.org/pub/pdb/validation_reports/me/1me4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1me3C  1f29S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |






















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological unit is a monomer |
-Components
| #1: Protein | Mass: 22715.133 Da / Num. of mol.: 1 / Fragment: CATALYTIC DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.)  References: UniProt: P25779, Hydrolases; Acting on peptide bonds (peptidases); Cysteine endopeptidases |
|---|---|
| #2: Chemical | ChemComp-T10 / [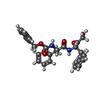  Mass: 460.522 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H28N2O5 Mass: 460.522 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H28N2O5 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 484 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 484 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.99 Å3/Da / Density % sol: 38.29 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 6.8 Details: 0.6 - 1.0 M Sodium Citrate, pH 6.8, VAPOR DIFFUSION, HANGING DROP, temperature 291K | ||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 18 ℃ / pH: 5.8 | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-1 / Wavelength: 0.98 Å / Beamline: BL9-1 / Wavelength: 0.98 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jun 8, 2002 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 1.1→30 Å / Num. obs: 51125 / % possible obs: 96.4 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 3.3 % / Rmerge(I) obs: 0.034 / Net I/σ(I): 26.7 |
| Reflection shell | Resolution: 1.2→1.23 Å / Redundancy: 2 % / Rmerge(I) obs: 0.13 / Mean I/σ(I) obs: 6.02 / Num. unique all: 3370 / % possible all: 91.1 |
| Reflection | *PLUS Lowest resolution: 30 Å / Num. obs: 53954 / Num. measured all: 440603 |
| Reflection shell | *PLUS % possible obs: 91.1 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1f29 Resolution: 1.2→30 Å / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.2→30 Å
| ||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | ||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 30 Å / Num. reflection Rfree: 1631 / % reflection Rfree: 10 % / Rfactor all: 0.096 / Rfactor Rfree: 0.13 / Rfactor Rwork: 0.0969 | ||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||
| Displacement parameters | *PLUS |
