 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bl4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | FKBP MUTANT F36V COMPLEXED WITH REMODELED SYNTHETIC LIGAND | ||||||
 Components Components | PROTEIN (FK506 BINDING PROTEIN) | ||||||
 Keywords Keywords | ISOMERASE / ROTAMASE | ||||||
| Function / homology |  Function and homology information Function and homology information: / extrinsic component of organelle membrane / macrolide binding / activin receptor binding / regulation of skeletal muscle contraction by regulation of release of sequestered calcium ion / cytoplasmic side of membrane / transforming growth factor beta receptor binding / TGFBR1 LBD Mutants in Cancer / type I transforming growth factor beta receptor binding / negative regulation of activin receptor signaling pathway ...: / extrinsic component of organelle membrane / macrolide binding / activin receptor binding / regulation of skeletal muscle contraction by regulation of release of sequestered calcium ion / cytoplasmic side of membrane / transforming growth factor beta receptor binding / TGFBR1 LBD Mutants in Cancer / type I transforming growth factor beta receptor binding / negative regulation of activin receptor signaling pathway / signaling receptor inhibitor activity / heart trabecula formation / I-SMAD binding / regulation of amyloid precursor protein catabolic process / terminal cisterna / ryanodine receptor complex / 'de novo' protein folding / ventricular cardiac muscle tissue morphogenesis / FK506 binding / TGF-beta receptor signaling activates SMADs / intracellular membrane-bounded organelle / regulation of ryanodine-sensitive calcium-release channel activity / mTORC1-mediated signalling / Calcineurin activates NFAT / regulation of immune response / heart morphogenesis / regulation of release of sequestered calcium ion into cytosol by sarcoplasmic reticulum / supramolecular fiber organization / regulation of cardiac muscle contraction by regulation of the release of sequestered calcium ion / sarcoplasmic reticulum membrane / T cell activation / peptidylprolyl isomerase / sarcoplasmic reticulum / peptidyl-prolyl cis-trans isomerase activity / TGF-beta receptor signaling in EMT (epithelial to mesenchymal transition) / calcium channel regulator activity / negative regulation of transforming growth factor beta receptor signaling pathway / protein maturation / Z disc / SARS-CoV-1 activates/modulates innate immune responses / regulation of protein localization / protein refolding / protein folding / amyloid fibril formation / Potential therapeutics for SARS / transmembrane transporter binding / positive regulation of canonical NF-kappaB signal transduction / membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Hatada, M.H. / Clackson, T. / Yang, W. / Rozamus, L.W. / Amara, J. / Rollins, C.T. / Stevenson, L.F. / Magari, S.R. / Wood, S.A. / Courage, N.L. ...Hatada, M.H. / Clackson, T. / Yang, W. / Rozamus, L.W. / Amara, J. / Rollins, C.T. / Stevenson, L.F. / Magari, S.R. / Wood, S.A. / Courage, N.L. / Lu, X. / Cerasoli Junior, F. / Gilman, M. / Holt, D. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 1998 Title: Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Authors: Clackson, T. / Yang, W. / Rozamus, L.W. / Hatada, M. / Amara, J.F. / Rollins, C.T. / Stevenson, L.F. / Magari, S.R. / Wood, S.A. / Courage, N.L. / Lu, X. / Cerasoli Jr., F. / Gilman, M. / Holt, D.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bl4.cif.gz | 58 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bl4.ent.gz | 41.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1bl4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bl/1bl4ftp://data.pdbj.org/pub/pdb/validation_reports/bl/1bl4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1fkfS S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 11788.465 Da / Num. of mol.: 2 / Mutation: F36V Source method: isolated from a genetically manipulated source Details: COMPLEXED WITH REDESIGNED COMPOUND / Source: (gene. exp.) Homo sapiens (human) / Production host:  References: UniProt: P62942, UniProt: P20071, peptidylprolyl isomerase #2: Chemical | 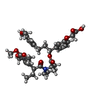  Mass: 693.780 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C38H47NO11 Mass: 693.780 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C38H47NO11#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 69 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 69 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 38 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 6 Details: VAPOR DIFFUSION IN HANGING DROPS WITH 40 MG/ML COMPLEX AND 1.2M AMMONIUM SULFATE, 0.1M SODIUM PHOSPHATE, PH 6.0 OVER RESERVOIRS OF 2.4 M AMMONIUM SULFATE, vapor diffusion - hanging drop | ||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Sep 15, 1996 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→20 Å / Num. obs: 16490 / % possible obs: 94.8 % / Observed criterion σ(I): 0 / Redundancy: 3.5 % / Biso Wilson estimate: 16.5 Å2 / Rmerge(I) obs: 0.063 |
| Reflection shell | Resolution: 1.9→2 Å / Redundancy: 3.5 % / Mean I/σ(I) obs: 3.9 / Rsym value: 0.16 / % possible all: 91 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1FKF Resolution: 1.9→10 Å / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.99 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file | Serial no: 1 / Param file: PARAM19X.PRO | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 10 Å / σ(F): 2 / % reflection Rfree: 10 % / Rfactor Rfree: 0.23 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.25 / % reflection Rfree: 10 % / Rfactor Rwork: 0.237 |
