National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
R00GM112892
United States
Other private
DFS-20-16 (Damon Runyon Cancer Research Fundation)
United States
National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
R01GM097312
United States
Howard Hughes Medical Institute (HHMI)
United States
Citation
Journal: EMBO J / Year: 2019 Title: Coupling of ATPase activity, microtubule binding, and mechanics in the dynein motor domain. Authors: Stefan Niekamp / Nicolas Coudray / Nan Zhang / Ronald D Vale / Gira Bhabha / Abstract: The movement of a molecular motor protein along a cytoskeletal track requires communication between enzymatic, polymer-binding, and mechanical elements. Such communication is particularly complex and ...The movement of a molecular motor protein along a cytoskeletal track requires communication between enzymatic, polymer-binding, and mechanical elements. Such communication is particularly complex and not well understood in the dynein motor, an ATPase that is comprised of a ring of six AAA domains, a large mechanical element (linker) spanning over the ring, and a microtubule-binding domain (MTBD) that is separated from the AAA ring by a ~ 135 Å coiled-coil stalk. We identified mutations in the stalk that disrupt directional motion, have microtubule-independent hyperactive ATPase activity, and nucleotide-independent low affinity for microtubules. Cryo-electron microscopy structures of a mutant that uncouples ATPase activity from directional movement reveal that nucleotide-dependent conformational changes occur normally in one-half of the AAA ring, but are disrupted in the other half. The large-scale linker conformational change observed in the wild-type protein is also inhibited, revealing that this conformational change is not required for ATP hydrolysis. These results demonstrate an essential role of the stalk in regulating motor activity and coupling conformational changes across the two halves of the AAA ring.
History
Deposition
Jan 3, 2019
-
Header (metadata) release
Mar 6, 2019
-
Map release
Mar 6, 2019
-
Update
Dec 11, 2019
-
Current status
Dec 11, 2019
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample
 Authors
Authors United States, 4 items
United States, 4 items  Citation
Citation Structure visualization
Structure visualization Movie viewer
Movie viewer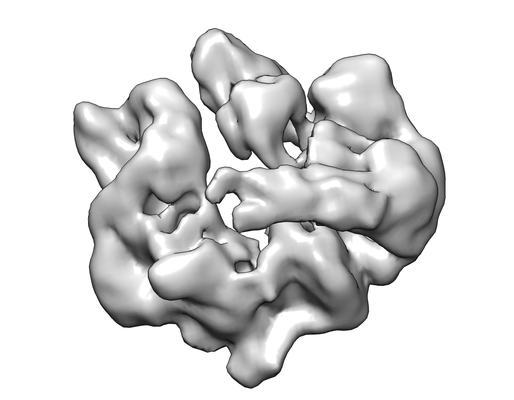
 Downloads & links
Downloads & links emd_9386.png
emd_9386.png http://ftp.pdbj.org/pub/emdb/structures/EMD-9386
http://ftp.pdbj.org/pub/emdb/structures/EMD-9386









 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)


















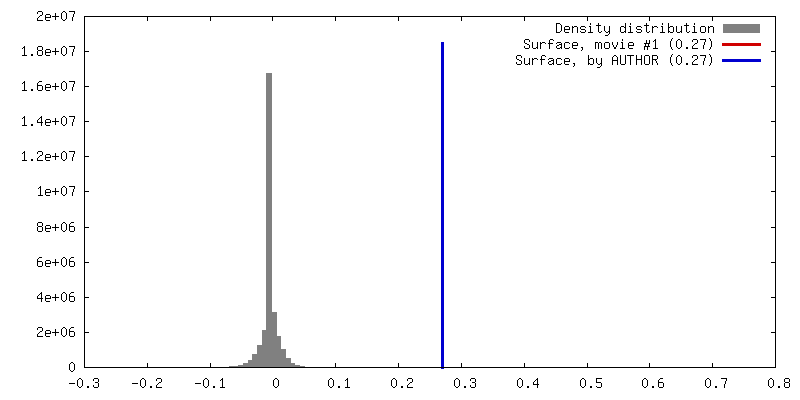

 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
