 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-6285 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Yeast V-ATPase state 2 | |||||||||
 マップデータ マップデータ | Yeast V-ATPase state 2 | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード | V-ATPase / V-type ATPase / vacuolar-type ATPase / yeast / Saccharomyces cerevisiae / hydrolase | |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報vacuole-mitochondrion membrane contact site / protein localization to vacuolar membrane / cellular response to alkaline pH / Insulin receptor recycling / Transferrin endocytosis and recycling / polyphosphate metabolic process / ROS and RNS production in phagocytes / Amino acids regulate mTORC1 / Golgi lumen acidification / proteasome storage granule assembly ...vacuole-mitochondrion membrane contact site / protein localization to vacuolar membrane / cellular response to alkaline pH / Insulin receptor recycling / Transferrin endocytosis and recycling / polyphosphate metabolic process / ROS and RNS production in phagocytes / Amino acids regulate mTORC1 / Golgi lumen acidification / proteasome storage granule assembly / vacuolar transport / vacuolar proton-transporting V-type ATPase, V1 domain / vacuolar proton-transporting V-type ATPase, V0 domain / endosomal lumen acidification / vacuole organization / protein targeting to vacuole / proton-transporting V-type ATPase complex / fungal-type vacuole / intron homing / pexophagy / vacuolar proton-transporting V-type ATPase complex / intein-mediated protein splicing / cellular hyperosmotic response / vacuolar acidification / fungal-type vacuole membrane / phosphatidylinositol-3,5-bisphosphate binding / proton transmembrane transporter activity / proton-transporting ATPase activity, rotational mechanism / intracellular copper ion homeostasis / H+-transporting two-sector ATPase / ATP metabolic process / Neutrophil degranulation / proton transmembrane transport / transmembrane transport / endocytosis / cytoplasmic stress granule / intracellular calcium ion homeostasis / ATPase binding / protein-containing complex assembly / endonuclease activity / 加水分解酵素; エステル加水分解酵素 / intracellular iron ion homeostasis / membrane raft / Golgi membrane / hydrolase activity / mRNA binding / ATP hydrolysis activity / DNA binding / ATP binding / membrane / cytoplasm 類似検索 - 分子機能 | |||||||||
| 生物種 |  | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 7.6 Å | |||||||||
 データ登録者 データ登録者 | Zhao J / Benlekbir S / Rubinstein JL | |||||||||
 引用 引用 | ジャーナル: Nature / 年: 2015 タイトル: Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. 著者: Jianhua Zhao / Samir Benlekbir / John L Rubinstein /  要旨: Eukaryotic vacuolar H(+)-ATPases (V-ATPases) are rotary enzymes that use energy from hydrolysis of ATP to ADP to pump protons across membranes and control the pH of many intracellular compartments. ...Eukaryotic vacuolar H(+)-ATPases (V-ATPases) are rotary enzymes that use energy from hydrolysis of ATP to ADP to pump protons across membranes and control the pH of many intracellular compartments. ATP hydrolysis in the soluble catalytic region of the enzyme is coupled to proton translocation through the membrane-bound region by rotation of a central rotor subcomplex, with peripheral stalks preventing the entire membrane-bound region from turning with the rotor. The eukaryotic V-ATPase is the most complex rotary ATPase: it has three peripheral stalks, a hetero-oligomeric proton-conducting proteolipid ring, several subunits not found in other rotary ATPases, and is regulated by reversible dissociation of its catalytic and proton-conducting regions. Studies of ATP synthases, V-ATPases, and bacterial/archaeal V/A-ATPases have suggested that flexibility is necessary for the catalytic mechanism of rotary ATPases, but the structures of different rotational states have never been observed experimentally. Here we use electron cryomicroscopy to obtain structures for three rotational states of the V-ATPase from the yeast Saccharomyces cerevisiae. The resulting series of structures shows ten proteolipid subunits in the c-ring, setting the ATP:H(+) ratio for proton pumping by the V-ATPase at 3:10, and reveals long and highly tilted transmembrane α-helices in the a-subunit that interact with the c-ring. The three different maps reveal the conformational changes that occur to couple rotation in the symmetry-mismatched soluble catalytic region to the membrane-bound proton-translocating region. Almost all of the subunits of the enzyme undergo conformational changes during the transitions between these three rotational states. The structures of these states provide direct evidence that deformation during rotation enables the smooth transmission of power through rotary ATPases. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_6285.map.gz | 57.3 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-6285-v30.xmlemd-6285.xml | 13.7 KB 13.7 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_6285_fsc.xml | 11 KB | 表示 | FSCデータファイル |
| 画像 | 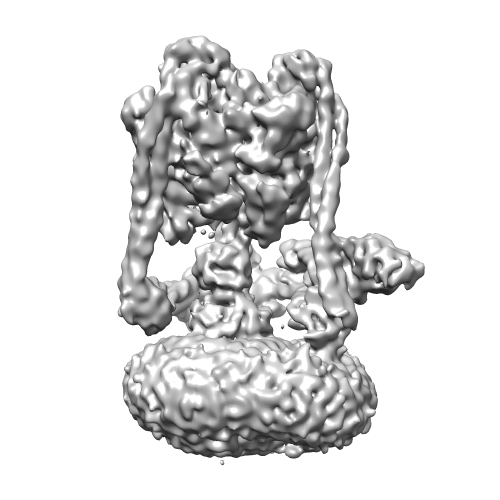 emd_6285.png emd_6285.png | 112.2 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-6285ftp://ftp.pdbj.org/pub/emdb/structures/EMD-6285 http://ftp.pdbj.org/pub/emdb/structures/EMD-6285ftp://ftp.pdbj.org/pub/emdb/structures/EMD-6285 | HTTPS FTP |
-関連構造データ












-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |












-マップ
| ファイル | ダウンロード / ファイル: emd_6285.map.gz / 形式: CCP4 / 大きさ: 62.5 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Yeast V-ATPase state 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
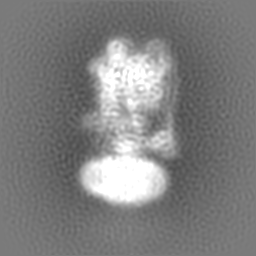
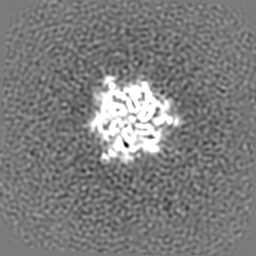
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.45 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : Vacuolar-type ATPase
| 全体 | 名称: Vacuolar-type ATPase |
|---|---|
| 要素 |
|
-超分子 #1000: Vacuolar-type ATPase
| 超分子 | 名称: Vacuolar-type ATPase / タイプ: sample / ID: 1000 / 詳細: Detergent solubilized protein complex 集合状態: A3B3CDE3FG3HadcXc'Yc''Z where X, Y, and Z indicate unknown stoichiometry and X+Y+Z=10 Number unique components: 1 |
|---|---|
| 分子量 | 実験値: 900 KDa / 理論値: 900 KDa / 手法: SDS-PAGE, size exclusion chromatography |
-分子 #1: Vacuolar-type ATPase
| 分子 | 名称: Vacuolar-type ATPase / タイプ: protein_or_peptide / ID: 1 / Name.synonym: V-ATPase, V-type ATPase / 詳細: Detergent-solubilized protein complex / コピー数: 1 / 集合状態: monomer / 組換発現: No / データベース: NCBI |
|---|---|
| 由来(天然) | 生物種: |
| 分子量 | 実験値: 900 KDa / 理論値: 900 KDa |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 10 mg/mL |
|---|---|
| 緩衝液 | pH: 7.4 詳細: 50 mM Tris-HCl, 150 mM NaCl, 0.02% w/v dodecylmaltoside |
| グリッド | 詳細: Homemade holey carbon on 400 square mesh Cu/Rh grid, glow-discharged 2 mins |
| 凍結 | 凍結剤: ETHANE-PROPANE MIXTURE / チャンバー内湿度: 100 % / チャンバー内温度: 77 K / 装置: FEI VITROBOT MARK III / 手法: Blot for 23 seconds before freezing. |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TECNAI F20 |
|---|---|
| アライメント法 | Legacy - 非点収差: Manually corrected by inspecting FFT. |
| 詳細 | K2 Summit in counting mode, 2 frames/s for 15 s |
| 日付 | 2013年10月19日 |
| 撮影 | カテゴリ: CCD / フィルム・検出器のモデル: GATAN K2 (4k x 4k) / 実像数: 3685 / 平均電子線量: 30 e/Å2 / ビット/ピクセル: 32 |
| 電子線 | 加速電圧: 200 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 倍率(補正後): 34483 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.0 mm / 最大 デフォーカス(公称値): 7.0 µm / 最小 デフォーカス(公称値): 1.5 µm / 倍率(公称値): 34483 |
| 試料ステージ | 試料ホルダーモデル: GATAN LIQUID NITROGEN |
| 実験機器 |  モデル: Tecnai F20 / 画像提供: FEI Company |
-画像解析
| 詳細 | Particles automatically selected using TMaCS and processed in Relion. |
|---|---|
| CTF補正 | 詳細: Each particle |
| 最終 再構成 | アルゴリズム: OTHER / 解像度のタイプ: BY AUTHOR / 解像度: 7.6 Å / 解像度の算出法: OTHER / ソフトウェア - 名称: Relion / 使用した粒子像数: 38347 |
| FSC曲線 (解像度の算出) |  |
-原子モデル構築 1
| 初期モデル | PDB ID: Chain - Chain ID: A |
|---|---|
| ソフトウェア | 名称: Chimera, MDFF |
| 詳細 | Rigid body fitting performed in Chimera first, followed by flexible fitting performed using Molecular Dynamics Flexible Fitting (MDFF). |
| 精密化 | 空間: REAL / プロトコル: FLEXIBLE FIT |
| 得られたモデル |  PDB-3j9u: |

-原子モデル構築 2
| 初期モデル | PDB ID: Chain - Chain ID: A |
|---|---|
| ソフトウェア | 名称: Chimera, MDFF |
| 詳細 | Rigid body fitting performed in Chimera first, followed by flexible fitting performed using Molecular Dynamics Flexible Fitting (MDFF). |
| 精密化 | 空間: REAL / プロトコル: FLEXIBLE FIT |
| 得られたモデル | PDB-3j9u: |

-原子モデル構築 3
| 初期モデル | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B |
|---|---|
| ソフトウェア | 名称: Chimera, MDFF |
| 詳細 | Rigid body fitting performed in Chimera first, followed by flexible fitting performed using Molecular Dynamics Flexible Fitting (MDFF). |
| 精密化 | 空間: REAL / プロトコル: FLEXIBLE FIT |
| 得られたモデル | PDB-3j9u: |

-原子モデル構築 4
| 初期モデル | PDB ID: Chain - #0 - Chain ID: E / Chain - #1 - Chain ID: G |
|---|---|
| ソフトウェア | 名称: Chimera, MDFF |
| 詳細 | Rigid body fitting performed in Chimera first, followed by flexible fitting performed using Molecular Dynamics Flexible Fitting (MDFF). |
| 精密化 | 空間: REAL / プロトコル: FLEXIBLE FIT |
| 得られたモデル | PDB-3j9u: |

