 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-51001: Circularly permuted lumazine synthase twisted tube with 28 Angstr... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Circularly permuted lumazine synthase twisted tube with 28 Angstrom gap between double strands | ||||||||||||
 Map data Map data | Combined half maps symmetrized and post-processed by DeepEMhancer with binary mask normalization mode using refinement mask. | ||||||||||||
 Sample Sample |
| ||||||||||||
 Keywords Keywords | protein cage / protein engineering / self-assembly / geometry / helical reconstruction / bionanotechnology / polymorphism / pentamer / DE NOVO PROTEIN | ||||||||||||
| Function / homology |  Function and homology information Function and homology information6,7-dimethyl-8-ribityllumazine synthase / 6,7-dimethyl-8-ribityllumazine synthase activity / riboflavin synthase complex / riboflavin biosynthetic process / cytosol / cytoplasm Similarity search - Function | ||||||||||||
| Biological species |   Aquifex aeolicus VF5 (bacteria) Aquifex aeolicus VF5 (bacteria) | ||||||||||||
| Method | helical reconstruction / cryo EM / Resolution: 3.1 Å | ||||||||||||
 Authors Authors | Koziej L / Azuma Y | ||||||||||||
| Funding support |  Poland, European Union, 3 items Poland, European Union, 3 items
| ||||||||||||
 Citation Citation | Journal: ACS Nano / Year: 2025 Title: Dynamic Assembly of Pentamer-Based Protein Nanotubes. Authors: Lukasz Koziej / Farzad Fatehi / Marta Aleksejczuk / Matthew J Byrne / Jonathan G Heddle / Reidun Twarock / Yusuke Azuma /  Abstract: Hollow proteinaceous particles are useful nanometric containers for delivery and catalysis. Understanding the molecular mechanisms and the geometrical theory behind the polymorphic protein assemblies ...Hollow proteinaceous particles are useful nanometric containers for delivery and catalysis. Understanding the molecular mechanisms and the geometrical theory behind the polymorphic protein assemblies provides a basis for designing ones with the desired morphology. As such, we found that a circularly permuted variant of a cage-forming enzyme, lumazine synthase, cpAaLS, assembles into a variety of hollow spherical and cylindrical structures in response to changes in ionic strength. Cryogenic electron microscopy revealed that these structures are composed entirely of pentameric subunits, and the dramatic cage-to-tube transformation is attributed to the moderately hindered 3-fold symmetry interaction and the imparted torsion angle of the building blocks, where both mechanisms are mediated by an α-helix domain that is untethered from the native position by circular permutation. Mathematical modeling suggests that the unique double- and triple-stranded helical arrangements of subunits are optimal tiling patterns, while different geometries should be possible by modulating the interaction angles of the pentagons. These structural insights into dynamic, pentamer-based protein cages and nanotubes afford guidelines for designing nanoarchitectures with customized morphology and assembly characteristics. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_51001.map.gz | 295.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-51001-v30.xmlemd-51001.xml | 31.7 KB 31.7 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_51001_fsc.xml | 14.5 KB | Display | FSC data file |
| Images |  emd_51001.png emd_51001.png | 177.8 KB | ||
| Masks | emd_51001_msk_1.mapemd_51001_msk_2.map | 325 MB 325 MB | Mask map | |
| Filedesc metadata | emd-51001.cif.gz | 7.4 KB | ||
| Others | emd_51001_additional_1.map.gzemd_51001_additional_2.map.gzemd_51001_additional_3.map.gzemd_51001_additional_4.map.gzemd_51001_half_map_1.map.gzemd_51001_half_map_2.map.gz | 156.5 MB 306.5 MB 88.6 MB 89.2 MB 300.9 MB 300.9 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-51001ftp://ftp.pdbj.org/pub/emdb/structures/EMD-51001 http://ftp.pdbj.org/pub/emdb/structures/EMD-51001ftp://ftp.pdbj.org/pub/emdb/structures/EMD-51001 | HTTPS FTP |
-Validation report
| Summary document | emd_51001_validation.pdf.gz | 165.6 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_51001_full_validation.pdf.gz | 165.2 KB | Display | |
| Data in XML | emd_51001_validation.xml.gz | 570 B | Display | |
| Data in CIF | emd_51001_validation.cif.gz | 483 B | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-51001ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-51001 | HTTPS FTP |
-Related structure data
| Related structure data |  9g3jMC  9g3hC  9g3iC  9g3mC  9g3nC  9g3oC  9g3pC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
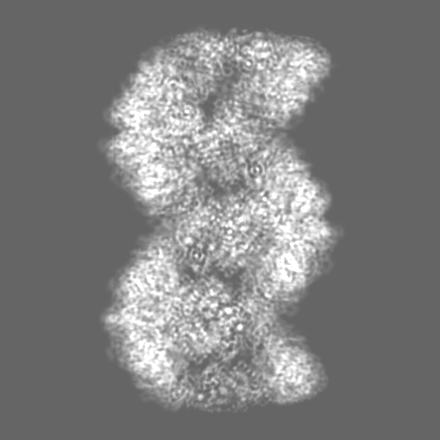
-Map
| File | Download / File: emd_51001.map.gz / Format: CCP4 / Size: 325 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Combined half maps symmetrized and post-processed by DeepEMhancer with binary mask normalization mode using refinement mask. | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.846 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Mask #1
| File | emd_51001_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Mask #2
| File | emd_51001_msk_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








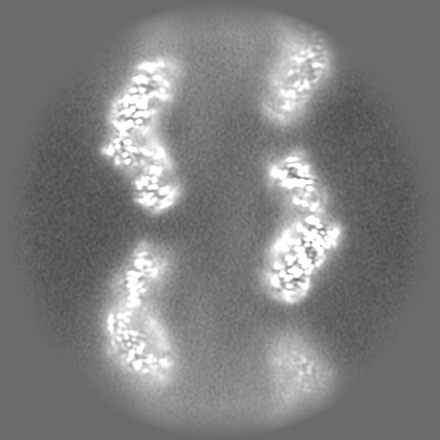
-Additional map: Map combined from 2 independent halves. Not post-processed...
| File | emd_51001_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Map combined from 2 independent halves. Not post-processed and not symmetrized map. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Additional map: Combined half maps post-processed using sharpening B factor 102.5.
| File | emd_51001_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Combined half maps post-processed using sharpening B factor 102.5. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Additional map: Combined half maps symmetrized and post-processed using sharpening...
| File | emd_51001_additional_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Combined half maps symmetrized and post-processed using sharpening B factor 102.5. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








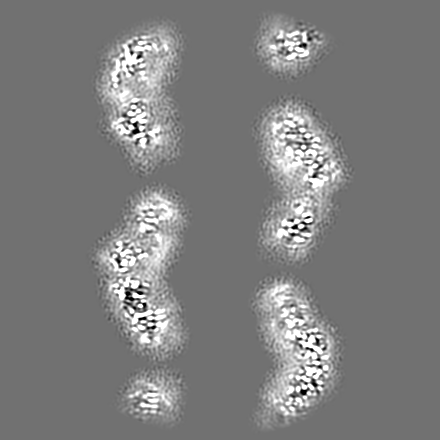
-Additional map: Combined half maps symmetrized and post-processed using sharpening...
| File | emd_51001_additional_4.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Combined half maps symmetrized and post-processed using sharpening B factor 102.5. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Half map: #1
| File | emd_51001_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #2
| File | emd_51001_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : Circularly permuted lumazine synthase twisted tube with 28 Angstr...
| Entire | Name: Circularly permuted lumazine synthase twisted tube with 28 Angstrom gap between double strands |
|---|---|
| Components |
|
-Supramolecule #1: Circularly permuted lumazine synthase twisted tube with 28 Angstr...
| Supramolecule | Name: Circularly permuted lumazine synthase twisted tube with 28 Angstrom gap between double strands type: complex / ID: 1 / Parent: 0 / Macromolecule list: all Details: Circularly permuted (119) variant of Aquifex aeolicus lumazine synthase with C37S and A85C mutations |
|---|---|
| Source (natural) | Organism: Aquifex aeolicus VF5 (bacteria) |
| Molecular weight | Theoretical: 600 kDa/nm |
-Macromolecule #1: 6,7-dimethyl-8-ribityllumazine synthase
| Macromolecule | Name: 6,7-dimethyl-8-ribityllumazine synthase / type: protein_or_peptide / ID: 1 / Details: cpAaLS(119, C37S, A85C),cpAaLS(119, C37S, A85C) / Number of copies: 100 / Enantiomer: LEVO / EC number: 6,7-dimethyl-8-ribityllumazine synthase |
|---|---|
| Source (natural) | Organism: Aquifex aeolicus VF5 (bacteria) |
| Molecular weight | Theoretical: 17.465934 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MTLEQAIERA GTKHGNKGWE AALSAIEMAN LFKSLRGTGG SGSSMEIYEG KLTAEGLRFG IVASRFNHAL VDRLVEGAID SIVRHGGRE EDITLVRVPG SWEIPVAAGE LARKEDIDAV IAIGVLIRGC TPHFDYIASE VSKGLANLSL ELRKPITFGV I TAD UniProtKB: 6,7-dimethyl-8-ribityllumazine synthase, 6,7-dimethyl-8-ribityllumazine synthase |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | helical reconstruction |
| Aggregation state | helical array |
-Sample preparation
| Concentration | 1 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7 Component:
| ||||||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 60 sec. / Pretreatment - Atmosphere: AIR | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 283 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | TFS KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 4092 pixel / Number grids imaged: 1 / Number real images: 12746 / Average electron dose: 42.44 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Calibrated magnification: 105000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 1.5 µm / Nominal defocus min: 0.9 µm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Initial model | PDB ID: Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Details | Initial fitting was done using ChimeraX. Flexible fitting was done using Isolde. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
| Output model | PDB-9g3j: |

