Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | PaaZ, bifunctional enzyme | |||||||||
 Map data Map data | Relion sharpened full map | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Hydrolase / dehydrogenase / Bi-functional enzyme | |||||||||
| Function / homology |  Function and homology information Function and homology information3-oxo-5,6-dehydrosuberyl-CoA semialdehyde dehydrogenase / oxepin-CoA hydrolase / hydrolase activity, acting on acid carbon-carbon bonds, in ketonic substances / ether hydrolase activity / oxidoreductase activity, acting on CH or CH2 groups, NAD or NADP as acceptor / phenylacetate catabolic process / oxidoreductase activity, acting on the aldehyde or oxo group of donors, NAD or NADP as acceptor / enoyl-CoA hydratase activity / identical protein binding Similarity search - Function | |||||||||
| Biological species |  | |||||||||
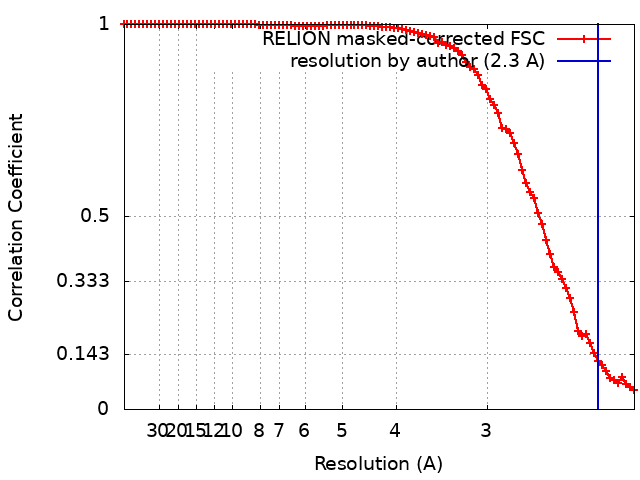
| Method | single particle reconstruction / cryo EM / Resolution: 2.3 Å | |||||||||
 Authors Authors | Yadav S / Vinothkumar KR | |||||||||
| Funding support |  India, 2 items India, 2 items
| |||||||||
 Citation Citation | Journal: Acta Crystallogr D Struct Biol / Year: 2024 Title: Factors affecting macromolecule orientations in thin films formed in cryo-EM. Authors: Swati Yadav / Kutti R Vinothkumar / Abstract: The formation of a vitrified thin film embedded with randomly oriented macromolecules is an essential prerequisite for cryogenic sample electron microscopy. Most commonly, this is achieved using the ...The formation of a vitrified thin film embedded with randomly oriented macromolecules is an essential prerequisite for cryogenic sample electron microscopy. Most commonly, this is achieved using the plunge-freeze method first described nearly 40 years ago. Although this is a robust method, the behaviour of different macromolecules shows great variation upon freezing and often needs to be optimized to obtain an isotropic, high-resolution reconstruction. For a macromolecule in such a film, the probability of encountering the air-water interface in the time between blotting and freezing and adopting preferred orientations is very high. 3D reconstruction using preferentially oriented particles often leads to anisotropic and uninterpretable maps. Currently, there are no general solutions to this prevalent issue, but several approaches largely focusing on sample preparation with the use of additives and novel grid modifications have been attempted. In this study, the effect of physical and chemical factors on the orientations of macromolecules was investigated through an analysis of selected well studied macromolecules, and important parameters that determine the behaviour of proteins on cryo-EM grids were revealed. These insights highlight the nature of the interactions that cause preferred orientations and can be utilized to systematically address orientation bias for any given macromolecule and to provide a framework to design small-molecule additives to enhance sample stability and behaviour. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_37866.map.gz | 59.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-37866-v30.xmlemd-37866.xml | 17.8 KB 17.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_37866_fsc.xml | 9 KB | Display | FSC data file |
| Images |  emd_37866.png emd_37866.png | 39.9 KB | ||
| Filedesc metadata | emd-37866.cif.gz | 6.3 KB | ||
| Others | emd_37866_half_map_1.map.gzemd_37866_half_map_2.map.gz | 49.5 MB 49.5 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-37866ftp://ftp.pdbj.org/pub/emdb/structures/EMD-37866 http://ftp.pdbj.org/pub/emdb/structures/EMD-37866ftp://ftp.pdbj.org/pub/emdb/structures/EMD-37866 | HTTPS FTP |
-Validation report
| Summary document | emd_37866_validation.pdf.gz | 990.9 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_37866_full_validation.pdf.gz | 990.5 KB | Display | |
| Data in XML | emd_37866_validation.xml.gz | 16.1 KB | Display | |
| Data in CIF | emd_37866_validation.cif.gz | 21.2 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-37866ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-37866 | HTTPS FTP |
-Related structure data
| Related structure data |  8wv6MC  8wv4C  8wv5C  8wzhC  8wziC  8wzjC  8wzkC  8wzmC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |









-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_37866.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Relion sharpened full map | ||||||||||||||||||||||||||||||||||||
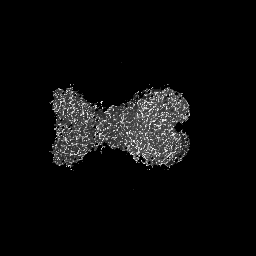
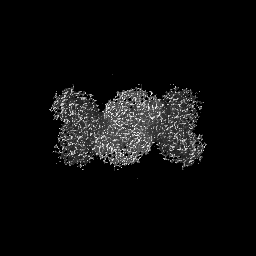
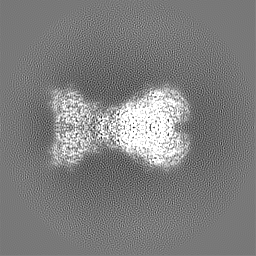
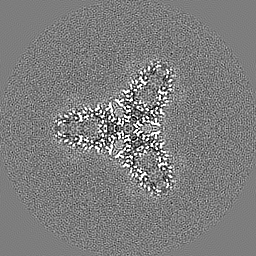
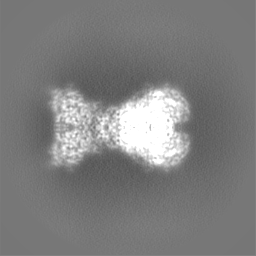
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.07 Å | ||||||||||||||||||||||||||||||||||||
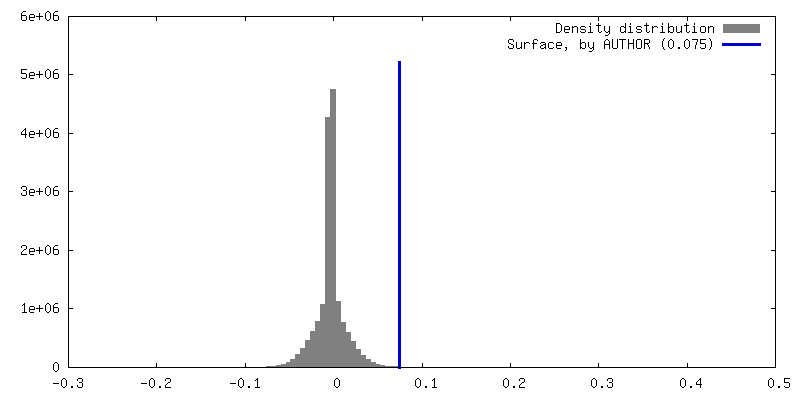
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)














-Supplemental data
-Half map: Relion sharpened half map
| File | emd_37866_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Relion sharpened half map | ||||||||||||
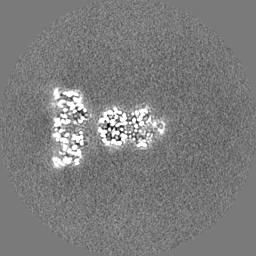
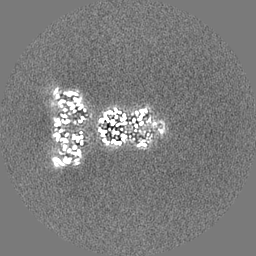
| Projections & Slices |
| ||||||||||||
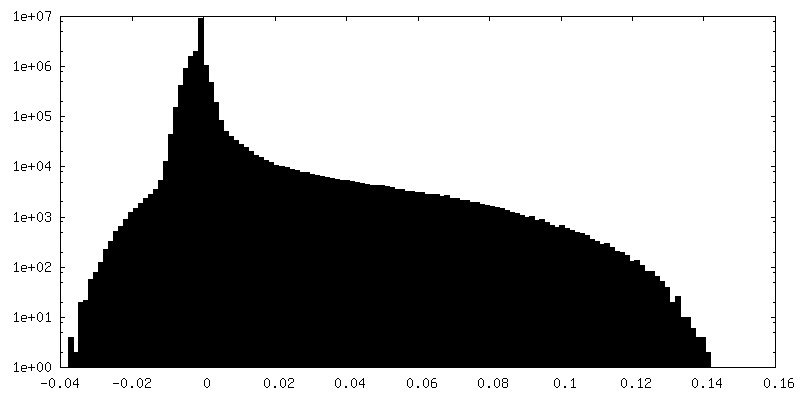
| Density Histograms |


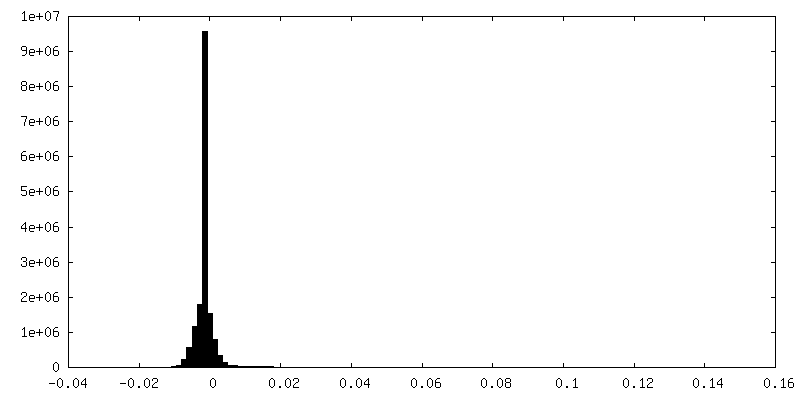
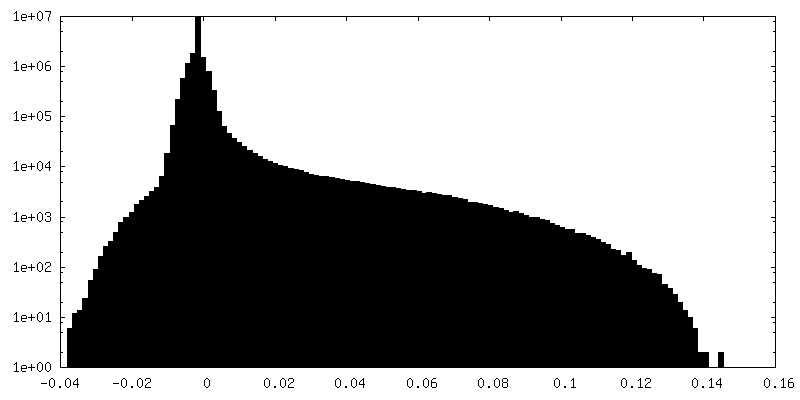
-Half map: Relion sharpened half map
| File | emd_37866_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Relion sharpened half map | ||||||||||||
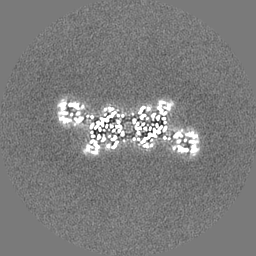
| Projections & Slices |
| ||||||||||||
| Density Histograms |





- Sample components
Sample components
-Entire : PaaZ, bifunctional enzyme
| Entire | Name: PaaZ, bifunctional enzyme |
|---|---|
| Components |
|
-Supramolecule #1: PaaZ, bifunctional enzyme
| Supramolecule | Name: PaaZ, bifunctional enzyme / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 438 KDa |
-Macromolecule #1: Bifunctional protein PaaZ
| Macromolecule | Name: Bifunctional protein PaaZ / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 73.969391 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MGHHHHHHQQ LASFLSGTWQ SGRGRSRLIH HAISGEALWE VTSEGLDMAA ARQFAIEKGA PALRAMTFIE RAAMLKAVAK HLLSEKERF YALSAQTGAT RADSWVDIEG GIGTLFTYAS LGSRELPDDT LWPEDELIPL SKEGGFAARH LLTSKSGVAV H INAFNFPC ...String: MGHHHHHHQQ LASFLSGTWQ SGRGRSRLIH HAISGEALWE VTSEGLDMAA ARQFAIEKGA PALRAMTFIE RAAMLKAVAK HLLSEKERF YALSAQTGAT RADSWVDIEG GIGTLFTYAS LGSRELPDDT LWPEDELIPL SKEGGFAARH LLTSKSGVAV H INAFNFPC WGMLEKLAPT WLGGMPAIIK PATATAQLTQ AMVKSIVDSG LVPEGAISLI CGSAGDLLDH LDSQDVVTFT GS AATGQML RVQPNIVAKS IPFTMEADSL NCCVLGEDVT PDQPEFALFI REVVREMTTK AGQKCTAIRR IIVPQALVNA VSD ALVARL QKVVVGDPAQ EGVKMGALVN AEQRADVQEK VNILLAAGCE IRLGGQADLS AAGAFFPPTL LYCPQPDETP AVHA TEAFG PVATLMPAQN QRHALQLACA GGGSLAGTLV TADPQIARQF IADAARTHGR IQILNEESAK ESTGHGSPLP QLVHG GPGR AGGGEELGGL RAVKHYMQRT AVQGSPTMLA AISKQWVRGA KVEEDRIHPF RKYFEELQPG DSLLTPRRTM TEADIV NFA CLSGDHFYAH MDKIAAAESI FGERVVHGYF VLSAAAGLFV DAGVGPVIAN YGLESLRFIE PVKPGDTIQV RLTCKRK TL KKQRSAEEKP TGVVEWAVEV FNQHQTPVAL YSILTLVARQ HGDFVD UniProtKB: Bifunctional protein PaaZ |
-Macromolecule #2: water
| Macromolecule | Name: water / type: ligand / ID: 2 / Number of copies: 288 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.8 mg/mL | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.4 Component:
| ||||||||
| Grid | Model: Quantifoil R0.6/1 / Material: GOLD / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 60 sec. / Pretreatment - Atmosphere: AIR | ||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV / Details: Blot force, 0. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Number grids imaged: 1 / Average exposure time: 60.0 sec. / Average electron dose: 25.0 e/Å2 Details: Images were collected in movie mode at 40 frames per second. Total of 25 frames were saved |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Calibrated magnification: 130841 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 3.0 µm / Nominal defocus min: 1.8 µm / Nominal magnification: 75000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |