 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-33945: Cryo-EM structure of MERS-CoV spike protein, One RBD-up conformation 3 -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of MERS-CoV spike protein, One RBD-up conformation 3 | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | MERS-CoV / Spike / Glycoprotein / Viral protein | |||||||||
| Biological species |  Human betacoronavirus 2c EMC/2012 Human betacoronavirus 2c EMC/2012 | |||||||||
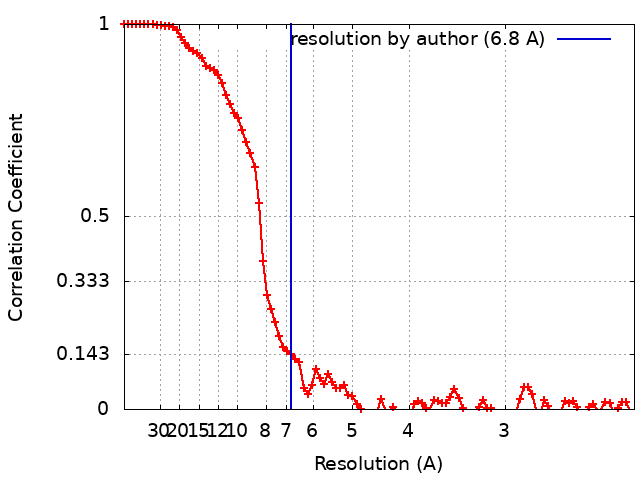
| Method | single particle reconstruction / cryo EM / Resolution: 6.8 Å | |||||||||
 Authors Authors | Hsu STD / Chang NE / Weng ZW / Yang TJ / Draczkowski P | |||||||||
| Funding support |  Taiwan, 1 items Taiwan, 1 items
| |||||||||
 Citation Citation | Journal: Cell / Year: 2024 Title: Rapid simulation of glycoprotein structures by grafting and steric exclusion of glycan conformer libraries. Authors: Yu-Xi Tsai / Ning-En Chang / Klaus Reuter / Hao-Ting Chang / Tzu-Jing Yang / Sören von Bülow / Vidhi Sehrawat / Noémie Zerrouki / Matthieu Tuffery / Michael Gecht / Isabell Louise ...Authors: Yu-Xi Tsai / Ning-En Chang / Klaus Reuter / Hao-Ting Chang / Tzu-Jing Yang / Sören von Bülow / Vidhi Sehrawat / Noémie Zerrouki / Matthieu Tuffery / Michael Gecht / Isabell Louise Grothaus / Lucio Colombi Ciacchi / Yong-Sheng Wang / Min-Feng Hsu / Kay-Hooi Khoo / Gerhard Hummer / Shang-Te Danny Hsu / Cyril Hanus / Mateusz Sikora /     Abstract: Most membrane proteins are modified by covalent addition of complex sugars through N- and O-glycosylation. Unlike proteins, glycans do not typically adopt specific secondary structures and remain ...Most membrane proteins are modified by covalent addition of complex sugars through N- and O-glycosylation. Unlike proteins, glycans do not typically adopt specific secondary structures and remain very mobile, shielding potentially large fractions of protein surface. High glycan conformational freedom hinders complete structural elucidation of glycoproteins. Computer simulations may be used to model glycosylated proteins but require hundreds of thousands of computing hours on supercomputers, thus limiting routine use. Here, we describe GlycoSHIELD, a reductionist method that can be implemented on personal computers to graft realistic ensembles of glycan conformers onto static protein structures in minutes. Using molecular dynamics simulation, small-angle X-ray scattering, cryoelectron microscopy, and mass spectrometry, we show that this open-access toolkit provides enhanced models of glycoprotein structures. Focusing on N-cadherin, human coronavirus spike proteins, and gamma-aminobutyric acid receptors, we show that GlycoSHIELD can shed light on the impact of glycans on the conformation and activity of complex glycoproteins. | |||||||||
| History |
|
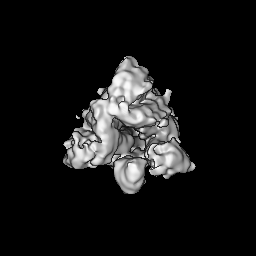
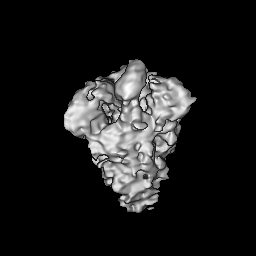
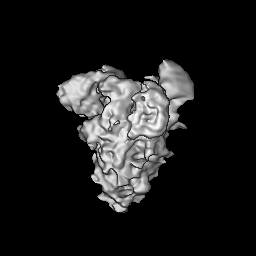
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_33945.map.gz | 30.8 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-33945-v30.xmlemd-33945.xml | 18.9 KB 18.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_33945_fsc.xml | 8.9 KB | Display | FSC data file |
| Images |  emd_33945.png emd_33945.png | 21.8 KB | ||
| Filedesc metadata | emd-33945.cif.gz | 5.8 KB | ||
| Others | emd_33945_additional_1.map.gzemd_33945_half_map_1.map.gzemd_33945_half_map_2.map.gz | 59.7 MB 59.3 MB 59.3 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-33945ftp://ftp.pdbj.org/pub/emdb/structures/EMD-33945 http://ftp.pdbj.org/pub/emdb/structures/EMD-33945ftp://ftp.pdbj.org/pub/emdb/structures/EMD-33945 | HTTPS FTP |
-Validation report
| Summary document | emd_33945_validation.pdf.gz | 753 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_33945_full_validation.pdf.gz | 752.6 KB | Display | |
| Data in XML | emd_33945_validation.xml.gz | 16.8 KB | Display | |
| Data in CIF | emd_33945_validation.cif.gz | 21.7 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-33945ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-33945 | HTTPS FTP |
-Related structure data















-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_33945.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
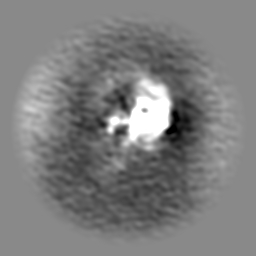
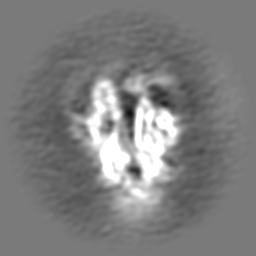
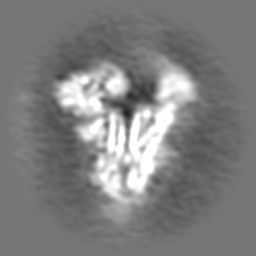
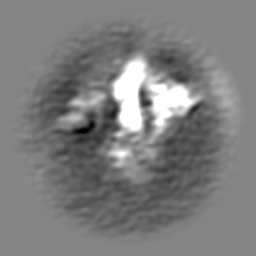
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size |
| ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)











-Supplemental data
-Additional map: #1
| File | emd_33945_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
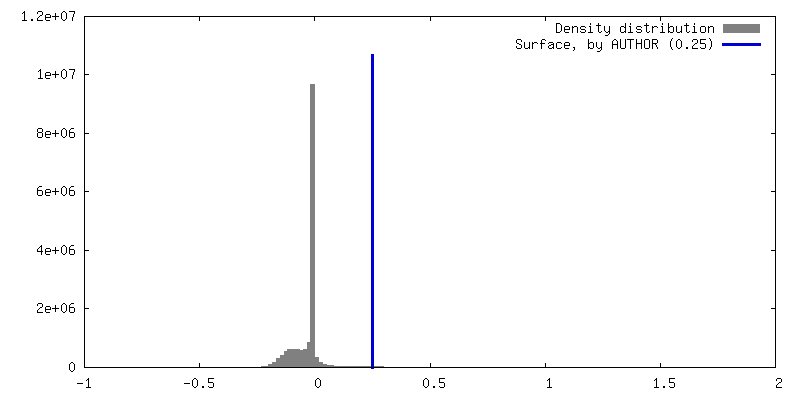
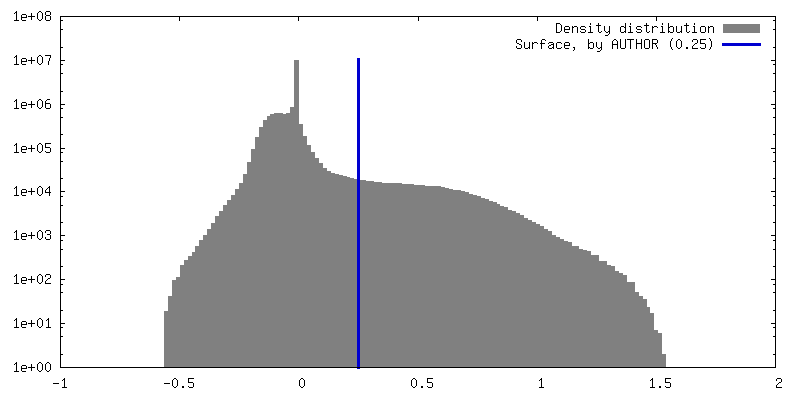
| Density Histograms |




-Half map: #1
| File | emd_33945_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |
- Sample components
Sample components
-Entire : recombinant MERS-CoV (betacoronavirus 2c EMC 2012) fm2P Spike
| Entire | Name: recombinant MERS-CoV (betacoronavirus 2c EMC 2012) fm2P Spike |
|---|---|
| Components |
|
-Supramolecule #1: recombinant MERS-CoV (betacoronavirus 2c EMC 2012) fm2P Spike
| Supramolecule | Name: recombinant MERS-CoV (betacoronavirus 2c EMC 2012) fm2P Spike type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Human betacoronavirus 2c EMC/2012 |
-Macromolecule #1: Spike glycoprotein
| Macromolecule | Name: Spike glycoprotein / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Human betacoronavirus 2c EMC/2012 |
| Recombinant expression | Organism:  Homo sapiens (human) Homo sapiens (human) |
| Sequence | String: MDSWFILVLL GSGLICVSAS YVDVGPDSVK SACIEVDIQQ TFFDKTWPRP IDVSKADGII YPQGRTYSNI TITYQGLFPY QGDHGDMYVY SAGHATGTTP QKLFVANYSQ DVKQFANGFV VRIGAAANST GTVIISPSTS ATIRKIYPAF MLGSSVGNFS DGKMGRFFNH ...String: MDSWFILVLL GSGLICVSAS YVDVGPDSVK SACIEVDIQQ TFFDKTWPRP IDVSKADGII YPQGRTYSNI TITYQGLFPY QGDHGDMYVY SAGHATGTTP QKLFVANYSQ DVKQFANGFV VRIGAAANST GTVIISPSTS ATIRKIYPAF MLGSSVGNFS DGKMGRFFNH TLVLLPDGCG TLLRAFYCIL EPRSGNHCPA GNSYTSFATY HTPATDCSDG NYNRNASLNS FKEYFNLRNC TFMYTYNITE DEILEWFGIT QTAQGVHLFS SRYVDLYGGN MFQFATLPVY DTIKYYSIIP HSIRSIQSDR KAWAAFYVYK LQPLTFLLDF SVDGYIRRAI DCGFNDLSQL HCSYESFDVE SGVYSVSSFE AKPSGSVVEQ AEGVECDFSP LLSGTPPQVY NFKRLVFTNC NYNLTKLLSL FSVNDFTCSQ ISPAAIASNC YSSLILDYFS YPLSMKSDLS VSSAGPISQF NYKQSFSNPT CLILATVPHN LTTITKPLKY SYINKCSRLL SDDRTEVPQL VNANQYSPCV SIVPSTVWED GDYYRKQLSP LEGGGWLVAS GSTVAMTEQL QMGFGITVQY GTDTNSVCPK LEFANDTKIA SQLGNCVEYS LYGVSGRGVF QNCTAVGVRQ QRFVYDAYQN LVGYYSDDGN YYCLRACVSV PVSVIYDKET KTHATLFGSV ACEHISSTMS QYSRSTRSML KRRDSTYGPL QTPVGCVLGL VNSSLFVEDC KLPLGQSLCA LPDTPSTLTP ASVGSVPGEM RLASIAFNHP IQVDQLNSSY FKLSIPTNFS FGVTQEYIQT TIQKVTVDCK QYVCNGFQKC EQLLREYGQF CSKINQALHG ANLRQDDSVR NLFASVKSSQ SSPIIPGFGG DFNLTLLEPV SISTGSRSAR SAIEDLLFDK VTIADPGYMQ GYDDCMQQGP ASARDLICAQ YVAGYKVLPP LMDVNMEAAY TSSLLGSIAG VGWTAGLSSF AAIPFAQSIF YRLNGVGITQ QVLSENQKLI ANKFNQALGA MQTGFTTTNE AFQKVQDAVN NNAQALSKLA SELSNTFGAI SASIGDIIQR LDPPEQDAQI DRLINGRLTT LNAFVAQQLV RSESAALSAQ LAKDKVNECV KAQSKRSGFC GQGTHIVSFV VNAPNGLYFM HVGYYPSNHI EVVSAYGLCD AANPTNCIAP VNGYFIKTNN TRIVDEWSYT GSSFYAPEPI TSLNTKYVAP QVTYQNISTN LPPPLLGNST GIDFQDELDE FFKNVSTSIP NFGSLTQINT TLLDLTYEML SLQQVVKALN ESYIDLKELG NYTYEFGSGG YIPEAPRDGQ AYVRKDGEWV LLSTFLKGQD NSADIQHSGR PLESRGPFEQ KLISEEDLNM HTGHHHHHH |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.5 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.6 Component:
| ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277.15 K / Instrument: FEI VITROBOT MARK IV Details: blot for 2.5 seconds before plunging; blot force: -1; waiting time: 30s.. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TALOS ARCTICA |
|---|---|
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Number grids imaged: 1 / Number real images: 2886 / Average electron dose: 40.6 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 1.7 µm / Nominal defocus min: 1.5 µm / Nominal magnification: 92000 |
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
|---|
