Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-38650: Additional map for SARS-CoV-2 Spike D614G variant, one RBD-up con... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Additional map for SARS-CoV-2 Spike D614G variant, one RBD-up conformation 1 (PDB ID: 7EAZ; EMD-31047). Map was generated from heterogeneous refinement with downsampling in CryoSPARC | |||||||||||||||
 Map data Map data | Additional map for SARS-CoV-2 Spike D614G variant, one RBD-up conformation 1 (PDB ID: 7EAZ; EMD-31047). Map was generated from heterogeneous refinement with downsampling | |||||||||||||||
 Sample Sample |
| |||||||||||||||
 Keywords Keywords | SARS-CoV-2 / Spike protein / VIRAL PROTEIN | |||||||||||||||
| Biological species |   Severe acute respiratory syndrome coronavirus 2 Severe acute respiratory syndrome coronavirus 2 | |||||||||||||||
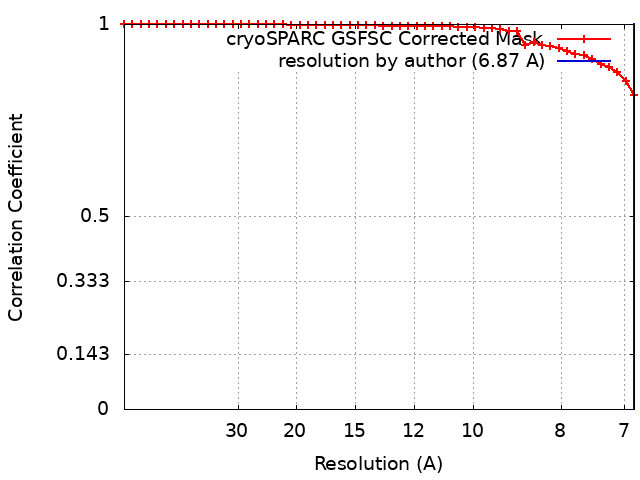
| Method | single particle reconstruction / cryo EM / Resolution: 6.87 Å | |||||||||||||||
 Authors Authors | Yang TJ / Yu PY / Hsu STD | |||||||||||||||
| Funding support |  Taiwan, 4 items Taiwan, 4 items
| |||||||||||||||
 Citation Citation | Journal: Cell / Year: 2024 Title: Rapid simulation of glycoprotein structures by grafting and steric exclusion of glycan conformer libraries. Authors: Yu-Xi Tsai / Ning-En Chang / Klaus Reuter / Hao-Ting Chang / Tzu-Jing Yang / Sören von Bülow / Vidhi Sehrawat / Noémie Zerrouki / Matthieu Tuffery / Michael Gecht / Isabell Louise ...Authors: Yu-Xi Tsai / Ning-En Chang / Klaus Reuter / Hao-Ting Chang / Tzu-Jing Yang / Sören von Bülow / Vidhi Sehrawat / Noémie Zerrouki / Matthieu Tuffery / Michael Gecht / Isabell Louise Grothaus / Lucio Colombi Ciacchi / Yong-Sheng Wang / Min-Feng Hsu / Kay-Hooi Khoo / Gerhard Hummer / Shang-Te Danny Hsu / Cyril Hanus / Mateusz Sikora /     Abstract: Most membrane proteins are modified by covalent addition of complex sugars through N- and O-glycosylation. Unlike proteins, glycans do not typically adopt specific secondary structures and remain ...Most membrane proteins are modified by covalent addition of complex sugars through N- and O-glycosylation. Unlike proteins, glycans do not typically adopt specific secondary structures and remain very mobile, shielding potentially large fractions of protein surface. High glycan conformational freedom hinders complete structural elucidation of glycoproteins. Computer simulations may be used to model glycosylated proteins but require hundreds of thousands of computing hours on supercomputers, thus limiting routine use. Here, we describe GlycoSHIELD, a reductionist method that can be implemented on personal computers to graft realistic ensembles of glycan conformers onto static protein structures in minutes. Using molecular dynamics simulation, small-angle X-ray scattering, cryoelectron microscopy, and mass spectrometry, we show that this open-access toolkit provides enhanced models of glycoprotein structures. Focusing on N-cadherin, human coronavirus spike proteins, and gamma-aminobutyric acid receptors, we show that GlycoSHIELD can shed light on the impact of glycans on the conformation and activity of complex glycoproteins. | |||||||||||||||
| History |
|
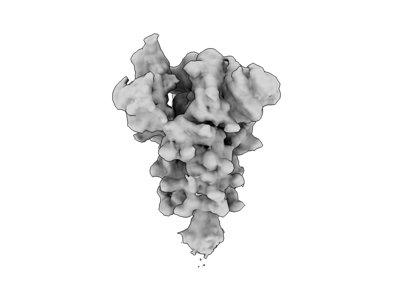
- Structure visualization
Structure visualization
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_38650.map.gz | 4.1 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-38650-v30.xmlemd-38650.xml | 17.1 KB 17.1 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_38650_fsc.xml | 4.2 KB | Display | FSC data file |
| Images |  emd_38650.png emd_38650.png | 31.5 KB | ||
| Filedesc metadata | emd-38650.cif.gz | 6 KB | ||
| Others | emd_38650_half_map_1.map.gzemd_38650_half_map_2.map.gz | 7.4 MB 7.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-38650ftp://ftp.pdbj.org/pub/emdb/structures/EMD-38650 http://ftp.pdbj.org/pub/emdb/structures/EMD-38650ftp://ftp.pdbj.org/pub/emdb/structures/EMD-38650 | HTTPS FTP |
-Related structure data
















-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_38650.map.gz / Format: CCP4 / Size: 8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Additional map for SARS-CoV-2 Spike D614G variant, one RBD-up conformation 1 (PDB ID: 7EAZ; EMD-31047). Map was generated from heterogeneous refinement with downsampling | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 3.3 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: Half map B
| File | emd_38650_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map B | ||||||||||||
| Projections & Slices |
| ||||||||||||
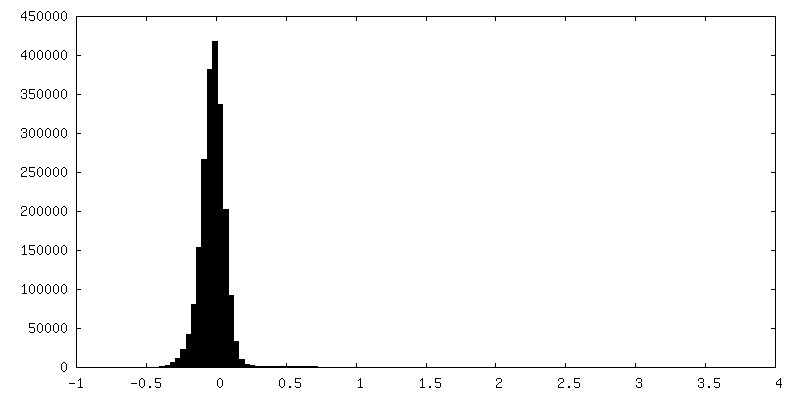
| Density Histograms |




- Sample components
Sample components
-Entire : SARS-CoV-2 spike glycoprotein
| Entire | Name: SARS-CoV-2 spike glycoprotein |
|---|---|
| Components |
|
-Supramolecule #1: SARS-CoV-2 spike glycoprotein
| Supramolecule | Name: SARS-CoV-2 spike glycoprotein / type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: all / Details: D614G variant, one RBD-up conformation 1 |
|---|---|
| Source (natural) | Organism: Severe acute respiratory syndrome coronavirus 2 |
| Molecular weight | Theoretical: 540 KDa |
-Macromolecule #1: Spike glycoprotein
| Macromolecule | Name: Spike glycoprotein / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Sequence | String: MFVFLVLLPL VSSQCVNLTT RTQLPPAYTN SFTRGVYYPD KVFRSSVLHS TQDLFLPFFS NVTWFHAIHV SGTNGTKRFD NPVLPFNDG VYFASTEKSN IIRGWIFGTT LDSKTQSLLI VNNATNVVIK VCEFQFCNDP FLGVYYHKNN KSWMESEFRV Y SSANNCTF ...String: MFVFLVLLPL VSSQCVNLTT RTQLPPAYTN SFTRGVYYPD KVFRSSVLHS TQDLFLPFFS NVTWFHAIHV SGTNGTKRFD NPVLPFNDG VYFASTEKSN IIRGWIFGTT LDSKTQSLLI VNNATNVVIK VCEFQFCNDP FLGVYYHKNN KSWMESEFRV Y SSANNCTF EYVSQPFLMD LEGKQGNFKN LREFVFKNID GYFKIYSKHT PINLVRDLPQ GFSALEPLVD LPIGINITRF QT LLALHRS YLTPGDSSSG WTAGAAAYYV GYLQPRTFLL KYNENGTITD AVDCALDPLS ETKCTLKSFT VEKGIYQTSN FRV QPTESI VRFPNITNLC PFGEVFNATR FASVYAWNRK RISNCVADYS VLYNSASFST FKCYGVSPTK LNDLCFTNVY ADSF VIRGD EVRQIAPGQT GKIADYNYKL PDDFTGCVIA WNSNNLDSKV GGNYNYLYRL FRKSNLKPFE RDISTEIYQA GSTPC NGVE GFNCYFPLQS YGFQPTNGVG YQPYRVVVLS FELLHAPATV CGPKKSTNLV KNKCVNFNFN GLTGTGVLTE SNKKFL PFQ QFGRDIADTT DAVRDPQTLE ILDITPCSFG GVSVITPGTN TSNQVAVLYQ GVNCTEVPVA IHADQLTPTW RVYSTGS NV FQTRAGCLIG AEHVNNSYEC DIPIGAGICA SYQTQTNSPG SASSVASQSI IAYTMSLGAE NSVAYSNNSI AIPTNFTI S VTTEILPVSM TKTSVDCTMY ICGDSTECSN LLLQYGSFCT QLNRALTGIA VEQDKNTQEV FAQVKQIYKT PPIKDFGGF NFSQILPDPS KPSKRSFIED LLFNKVTLAD AGFIKQYGDC LGDIAARDLI CAQKFNGLTV LPPLLTDEMI AQYTSALLAG TITSGWTFG AGAALQIPFA MQMAYRFNGI GVTQNVLYEN QKLIANQFNS AIGKIQDSLS STASALGKLQ DVVNQNAQAL N TLVKQLSS NFGAISSVLN DILSRLDPPE AEVQIDRLIT GRLQSLQTYV TQQLIRAAEI RASANLAATK MSECVLGQSK RV DFCGKGY HLMSFPQSAP HGVVFLHVTY VPAQEKNFTT APAICHDGKA HFPREGVFVS NGTHWFVTQR NFYEPQIITT DNT FVSGNC DVVIGIVNNT VYDPLQPELD SFKEELDKYF KNHTSPDVDL GDISGINASV VNIQKEIDRL NEVAKNLNES LIDL QELGK YEQEFGSGGY IPEAPRDGQA YVRKDGEWVL LSTFLKGQDN SADIQHSGRP LESRGPFEQK LISEEDLNMH TGHHH HHH |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1 mg/mL | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.6 Component:
| ||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV Details: blot for 2.5 seconds before plunging; blot force: 0; waiting time: 30s. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 1.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.6 µm / Nominal defocus min: 0.8 µm |
| Sample stage | Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |