 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3348 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | cryo-EM structure of T20S proteasome | |||||||||
 Map data Map data | cryo-EM reconstruction of T20S proteasome | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | proteasome / 20S | |||||||||
| Biological species |   Thermoplasma acidophilum (acidophilic) Thermoplasma acidophilum (acidophilic) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.1 Å | |||||||||
 Authors Authors | Danev R / Baumeister W | |||||||||
 Citation Citation | Journal: Elife / Year: 2016 Title: Cryo-EM single particle analysis with the Volta phase plate. Authors: Radostin Danev / Wolfgang Baumeister /  Abstract: We present a method for in-focus data acquisition with a phase plate that enables near-atomic resolution single particle reconstructions. Accurate focusing is the determining factor for obtaining ...We present a method for in-focus data acquisition with a phase plate that enables near-atomic resolution single particle reconstructions. Accurate focusing is the determining factor for obtaining high quality data. A double-area focusing strategy was implemented in order to achieve the required precision. With this approach we obtained a 3.2 Å resolution reconstruction of the Thermoplasma acidophilum 20S proteasome. The phase plate matches or slightly exceeds the performance of the conventional defocus approach. Spherical aberration becomes a limiting factor for achieving resolutions below 3 Å with in-focus phase plate images. The phase plate could enable single particle analysis of challenging samples in terms of small size, heterogeneity and flexibility that are difficult to solve by the conventional defocus approach. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3348.map.gz | 19.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3348-v30.xmlemd-3348.xml | 11.2 KB 11.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_3348_fsc.xml | 7.3 KB | Display | FSC data file |
| Images | 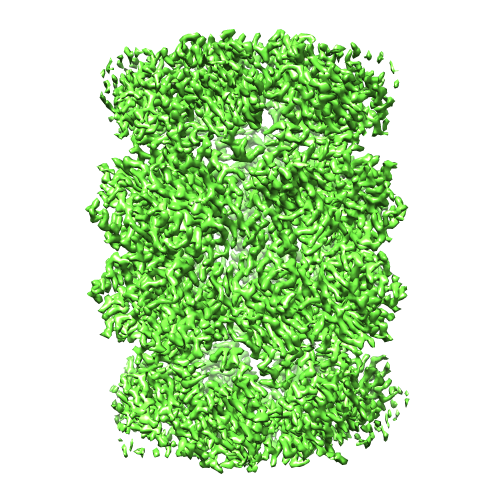 emd_3348.png emd_3348.png | 294.9 KB | ||
| Masks | emd_3348_msk_1.map | 20.8 MB | Mask map | |
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3348ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3348 http://ftp.pdbj.org/pub/emdb/structures/EMD-3348ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3348 | HTTPS FTP |
-Validation report
| Summary document | emd_3348_validation.pdf.gz | 371.9 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_3348_full_validation.pdf.gz | 371 KB | Display | |
| Data in XML | emd_3348_validation.xml.gz | 9.2 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-3348ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-3348 | HTTPS FTP |
-Related structure data
| Related structure data |  3347C C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10058 (Title: Cryo-EM dataset of T20S proteasome / Data size: 93.3 Data #1: Cryo-EM in-focus unaligned multi-frame micrographs of T20S proteasome [micrographs - multiframe]) |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_3348.map.gz / Format: CCP4 / Size: 20.3 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | cryo-EM reconstruction of T20S proteasome | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.35 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Segmentation: the mask was automatically generated by Relion during postprocessing.
| Annotation | the mask was automatically generated by Relion during postprocessing. | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| File | emd_3348_msk_1.map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : Thermoplasma acidophilum 20S proteasome
| Entire | Name: Thermoplasma acidophilum 20S proteasome |
|---|---|
| Components |
|
-Supramolecule #1000: Thermoplasma acidophilum 20S proteasome
| Supramolecule | Name: Thermoplasma acidophilum 20S proteasome / type: sample / ID: 1000 / Oligomeric state: 28-mer / Number unique components: 1 |
|---|---|
| Molecular weight | Theoretical: 700 KDa |
-Macromolecule #1: 20S proteasome
| Macromolecule | Name: 20S proteasome / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Oligomeric state: 28-mer / Recombinant expression: Yes |
|---|---|
| Source (natural) | Organism: Thermoplasma acidophilum (acidophilic) / synonym: Thermoplasma |
| Molecular weight | Theoretical: 700 KDa |
| Recombinant expression | Organism:  |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.5 mg/mL |
|---|---|
| Buffer | pH: 7 / Details: 25 mM Tris-HCl |
| Grid | Details: 200 mesh Copper Quantifoil R 1.2/1.3 |
| Vitrification | Cryogen name: ETHANE-PROPANE MIXTURE / Chamber humidity: 95 % / Chamber temperature: 80 K / Instrument: FEI VITROBOT MARK III Method: 3 ul of sample, blot time 5 s, chamber temperature 20 degC |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Average: 80 K |
| Alignment procedure | Legacy - Astigmatism: astigmatism was corrected manually at the magnification used for data acquisition. |
| Specialist optics | Energy filter - Name: Gatan Quantum / Energy filter - Lower energy threshold: 0.0 eV / Energy filter - Upper energy threshold: 10.0 eV |
| Date | Jul 21, 2015 |
| Image recording | Category: CCD / Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Digitization - Sampling interval: 5 µm / Number real images: 293 / Average electron dose: 30 e/Å2 Details: every image is the average of 12 frames on the direct detector |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 37037 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 1.7 µm / Nominal defocus min: 0.8 µm / Nominal magnification: 37000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Details | the particles were semi-automatically selected with EMAN2 e2boxer |
|---|---|
| CTF correction | Details: each micrograph |
| Final reconstruction | Applied symmetry - Point group: D7 (2x7 fold dihedral) / Resolution.type: BY AUTHOR / Resolution: 3.1 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: EMAN2, Relion, ctffind4 / Number images used: 35717 |
| FSC plot (resolution estimation) |  |
