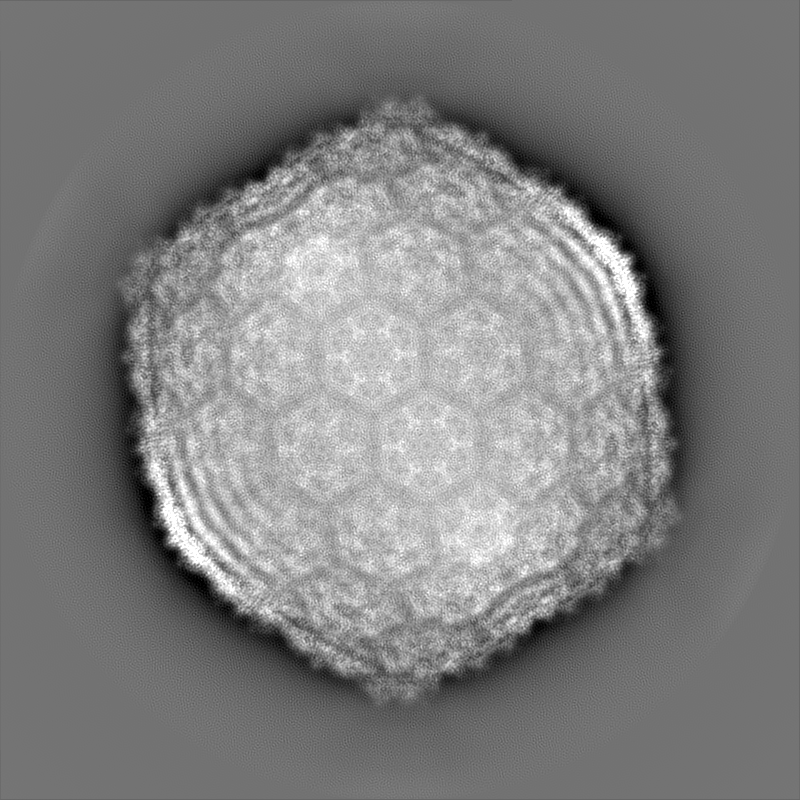
Journal: J Virol / Year: 2021 Title: Capsid Structure of Cyanophage A-1(L). Authors: Ning Cui / Feng Yang / Jun-Tao Zhang / Hui Sun / Yu Chen / Rong-Cheng Yu / Zhi-Peng Chen / Yong-Liang Jiang / Shu-Jing Han / Xudong Xu / Qiong Li / Cong-Zhao Zhou / Abstract: A-1(L) is a freshwater cyanophage with a contractile tail that specifically infects sp. PCC 7120, one of the model strains for molecular studies of cyanobacteria. Although isolated for half a ...A-1(L) is a freshwater cyanophage with a contractile tail that specifically infects sp. PCC 7120, one of the model strains for molecular studies of cyanobacteria. Although isolated for half a century, its structure remains unknown, which limits our understanding on the interplay between A-1(L) and its host. Here we report the 3.35 Å cryo-EM structure of A-1(L) capsid, representing the first near-atomic resolution structure of a phage capsid with a T number of 9. The major capsid gp4 proteins assemble into 91 capsomers, including 80 hexons: 20 at the center of the facet and 60 at the facet edge, in addition to 11 identical pentons. These capsomers further assemble into the icosahedral capsid, via gradually increasing curvatures. Different from the previously reported capsids of known-structure, A-1(L) adopts a noncovalent chainmail structure of capsid stabilized by two kinds of mortise-and-tenon inter-capsomer interactions: a three-layered interface at the pseudo 3-fold axis combined with the complementarity in shape and electrostatic potential around the 2-fold axis. This unique capsomer construction enables A-1(L) to possess a rigid capsid, which is solely composed of the major capsid proteins with an HK97 fold. Cyanobacteria are the most abundant photosynthetic bacteria, contributing significantly to the biomass production, O generation, and CO consumption on our planet. Their community structure and homeostasis in natural aquatic ecosystems are largely regulated by the corresponding cyanophages. In this study, we solved the structure of cyanophage A-1(L) capsid at near-atomic resolution and revealed a unique capsid construction. This capsid structure provides the molecular details for better understanding the assembly of A-1(L), and a structural platform for future investigation and application of A-1(L) in combination with its host sp. PCC 7120. As the first isolated freshwater cyanophage that infects the genetically tractable model cyanobacterium, A-1(L) should become an ideal template for the genetic engineering and synthetic biology studies.
History
Deposition
Jun 15, 2021
-
Header (metadata) release
Sep 29, 2021
-
Map release
Sep 29, 2021
-
Update
Jun 12, 2024
-
Current status
Jun 12, 2024
Processing site: PDBj / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information Nostoc phage A1 (virus)
Nostoc phage A1 (virus) Authors
Authors China, 1 items
China, 1 items  Citation
Citation Structure visualization
Structure visualization
 Downloads & links
Downloads & links emd_31431.png
emd_31431.png http://ftp.pdbj.org/pub/emdb/structures/EMD-31431
http://ftp.pdbj.org/pub/emdb/structures/EMD-31431










 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















 Sample components
Sample components Nostoc sp. PCC 7120 = FACHB-418 (bacteria)
Nostoc sp. PCC 7120 = FACHB-418 (bacteria) Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
