 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-27842: Structure of Lates calcarifer Twinkle helicase with ATP and DNA -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of Lates calcarifer Twinkle helicase with ATP and DNA | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Helicase Mitochondrion / REPLICATION-DNA complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationmitochondrial DNA replication / single-stranded DNA binding / 5'-3' DNA helicase activity / mitochondrion / ATP binding Similarity search - Function | |||||||||
| Biological species |  Lates calcarifer (barramundi perch) / synthetic construct (others) Lates calcarifer (barramundi perch) / synthetic construct (others) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.51 Å | |||||||||
 Authors Authors | Gao Y / Li Z | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: Nucleic Acids Res / Year: 2022 Title: Structural and dynamic basis of DNA capture and translocation by mitochondrial Twinkle helicase. Authors: Zhuo Li / Parminder Kaur / Chen-Yu Lo / Neil Chopra / Jamie Smith / Hong Wang / Yang Gao / Abstract: Twinkle is a mitochondrial replicative helicase which can self-load onto and unwind mitochondrial DNA. Nearly 60 mutations on Twinkle have been linked to human mitochondrial diseases. Using cryo- ...Twinkle is a mitochondrial replicative helicase which can self-load onto and unwind mitochondrial DNA. Nearly 60 mutations on Twinkle have been linked to human mitochondrial diseases. Using cryo-electron microscopy (cryo-EM) and high-speed atomic force microscopy (HS-AFM), we obtained the atomic-resolution structure of a vertebrate Twinkle homolog with DNA and captured in real-time how Twinkle is self-loaded onto DNA. Our data highlight the important role of the non-catalytic N-terminal domain of Twinkle. The N-terminal domain directly contacts the C-terminal helicase domain, and the contact interface is a hotspot for disease-related mutations. Mutations at the interface destabilize Twinkle hexamer and reduce helicase activity. With HS-AFM, we observed that a highly dynamic Twinkle domain, which is likely to be the N-terminal domain, can protrude ∼5 nm to transiently capture nearby DNA and initialize Twinkle loading onto DNA. Moreover, structural analysis and subunit doping experiments suggest that Twinkle hydrolyzes ATP stochastically, which is distinct from related helicases from bacteriophages. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_27842.map.gz | 118.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-27842-v30.xmlemd-27842.xml | 17.5 KB 17.5 KB | Display Display | EMDB header |
| Images |  emd_27842.png emd_27842.png | 127.1 KB | ||
| Filedesc metadata | emd-27842.cif.gz | 5.9 KB | ||
| Others | emd_27842_half_map_1.map.gzemd_27842_half_map_2.map.gz | 116.1 MB 116.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-27842ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27842 http://ftp.pdbj.org/pub/emdb/structures/EMD-27842ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27842 | HTTPS FTP |
-Validation report
| Summary document | emd_27842_validation.pdf.gz | 1.2 MB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_27842_full_validation.pdf.gz | 1.2 MB | Display | |
| Data in XML | emd_27842_validation.xml.gz | 14.1 KB | Display | |
| Data in CIF | emd_27842_validation.cif.gz | 16.5 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-27842ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-27842 | HTTPS FTP |
-Related structure data
| Related structure data |  8e2lMC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |



-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_27842.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.07 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: #2
| File | emd_27842_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
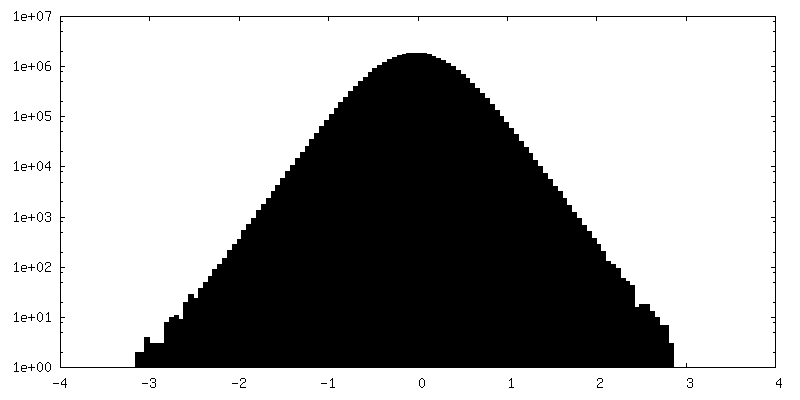
| Density Histograms |

-Half map: #1
| File | emd_27842_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |




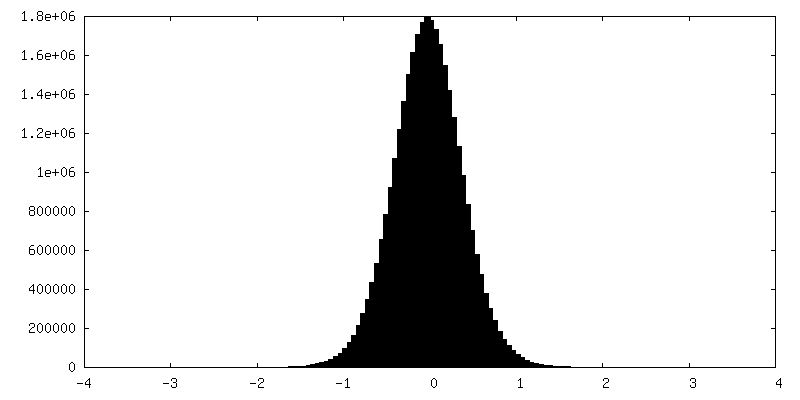
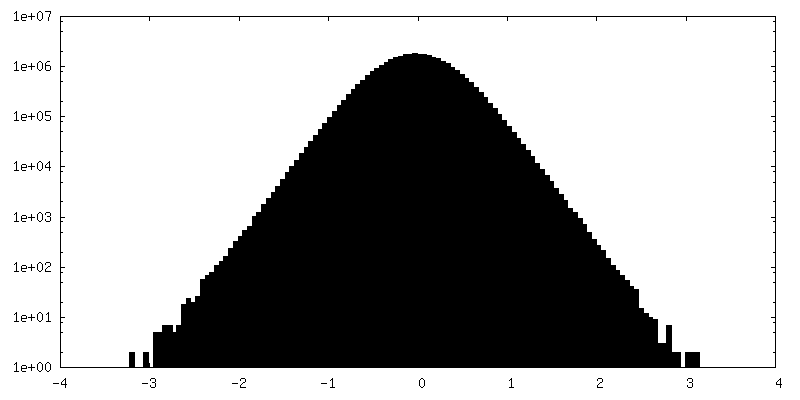
- Sample components
Sample components
-Entire : Twinkle protein, mitochondrial + DNA
| Entire | Name: Twinkle protein, mitochondrial + DNA |
|---|---|
| Components |
|
-Supramolecule #1: Twinkle protein, mitochondrial + DNA
| Supramolecule | Name: Twinkle protein, mitochondrial + DNA / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#2 |
|---|---|
| Source (natural) | Organism: Lates calcarifer (barramundi perch) |
-Macromolecule #1: Twinkle mtDNA helicase
| Macromolecule | Name: Twinkle mtDNA helicase / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Lates calcarifer (barramundi perch) |
| Molecular weight | Theoretical: 61.29816 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: MQKEEQDFLS PHVMLGYPES LDEQEEGERE LREVQRIWSS AVPFNDLPED EAQLIKTMFQ ITKVSNATLK KFGVRLFKPT KSLVFPWFA GPDSSLKGLK LLSAQNTDTE KVTYNEATVP KISSYYNLFG LTLVGRMDSE VVLTGHELDT LAVSQATGLP S VALPRGVS ...String: MQKEEQDFLS PHVMLGYPES LDEQEEGERE LREVQRIWSS AVPFNDLPED EAQLIKTMFQ ITKVSNATLK KFGVRLFKPT KSLVFPWFA GPDSSLKGLK LLSAQNTDTE KVTYNEATVP KISSYYNLFG LTLVGRMDSE VVLTGHELDT LAVSQATGLP S VALPRGVS CLPPMLLPYL EQFKRVTLWL GHDIRSWEAS KIFSRKLGLR RCSLVRPGED RPCPLEALAR GKNLSRIIKT SI PAAHKSI VSFKQLREDV YGELLNTEQV AGVKWTRFPE LNRILKGHRK GELTVFTGPT GSGKTTFISE VALDLCIQGV NTL WGSFQI NNVRLAKIML TQFAMQRLEE NLEQYDFWAD KFEELPLYFM TFHGQQNIKT VLDTMQHAVY LYDINHVIID NLQF MMGQE NLSIDKYAVQ DHIIGAFRKF ATNTSCHVTL IIHPRKEEDD RELQTASIFG SAKASQEADN VLILQEKKLV TCPGR RSLQ VTKNRFDGDV GIFPLDFIKS SLTFSAPIKG KVKLRKVSTK PENEEVGGER GGSEEGRG UniProtKB: Twinkle mtDNA helicase |
-Macromolecule #2: DNA (5'-D(P*TP*TP*TP*TP*TP*TP*TP*TP*TP*TP*TP*T)-3')
| Macromolecule | Name: DNA (5'-D(P*TP*TP*TP*TP*TP*TP*TP*TP*TP*TP*TP*T)-3') / type: dna / ID: 2 / Number of copies: 1 / Classification: DNA |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Theoretical: 4.517935 KDa |
| Sequence | String: (DT)(DT)(DT)(DT)(DT)(DT)(DT)(DT)(DT)(DT) (DT)(DT)(DT)(DT)(DT) |
-Macromolecule #3: ADENOSINE-5'-TRIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-TRIPHOSPHATE / type: ligand / ID: 3 / Number of copies: 5 / Formula: ATP |
|---|---|
| Molecular weight | Theoretical: 507.181 Da |
| Chemical component information |  ChemComp-ATP: |
-Macromolecule #4: MAGNESIUM ION
| Macromolecule | Name: MAGNESIUM ION / type: ligand / ID: 4 / Number of copies: 5 / Formula: MG |
|---|---|
| Molecular weight | Theoretical: 24.305 Da |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.5 mg/mL |
|---|---|
| Buffer | pH: 8 |
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 300 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 60 sec. |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 298 K / Instrument: FEI VITROBOT MARK I |
| Details | 50 mM Tris (pH 8.0), 150 mM KCl, 3 mM DTT, 1 mM ATP, and 2 mM MgCl2 |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Average electron dose: 49.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 3.0 µm / Nominal defocus min: 0.6 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Startup model | Type of model: NONE |
|---|---|
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Resolution.type: BY AUTHOR / Resolution: 3.51 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: cryoSPARC / Number images used: 44814 |
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: cryoSPARC |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: cryoSPARC |
-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: AB INITIO MODEL |
|---|---|
| Output model | PDB-8e2l: |
