 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-25185: Ab initio structure of proteinase K from electron-counted MicroED data -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Ab initio structure of proteinase K from electron-counted MicroED data | |||||||||
 Map data Map data | 1.5A resolution 2mFo-dFc MicroED map of proteinase K determined ab initio by electron-counted data | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Hydrolase | |||||||||
| Function / homology |  Function and homology information Function and homology informationpeptidase K / serine-type endopeptidase activity / proteolysis / extracellular region / metal ion binding Similarity search - Function | |||||||||
| Biological species |  Parengyodontium album (fungus) Parengyodontium album (fungus) | |||||||||
| Method | electron crystallography / cryo EM | |||||||||
 Authors Authors | Martynowycz MW / Clabbers MTB | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: Nat Methods / Year: 2022 Title: Ab initio phasing macromolecular structures using electron-counted MicroED data. Authors: Michael W Martynowycz / Max T B Clabbers / Johan Hattne / Tamir Gonen / Abstract: Structures of two globular proteins were determined ab initio using microcrystal electron diffraction (MicroED) data that were collected on a direct electron detector in counting mode. Microcrystals ...Structures of two globular proteins were determined ab initio using microcrystal electron diffraction (MicroED) data that were collected on a direct electron detector in counting mode. Microcrystals were identified using a scanning electron microscope (SEM) and thinned with a focused ion beam (FIB) to produce crystalline lamellae of ideal thickness. Continuous-rotation data were collected using an ultra-low exposure rate to enable electron counting in diffraction. For the first sample, triclinic lysozyme extending to a resolution of 0.87 Å, an ideal helical fragment of only three alanine residues provided initial phases. These phases were improved using density modification, allowing the entire atomic structure to be built automatically. A similar approach was successful on a second macromolecular sample, proteinase K, which is much larger and diffracted to a resolution of 1.5 Å. These results demonstrate that macromolecules can be determined to sub-ångström resolution by MicroED and that ab initio phasing can be successfully applied to counting data. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_25185.map.gz | 9.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-25185-v30.xmlemd-25185.xml | 14.9 KB 14.9 KB | Display Display | EMDB header |
| Images |  emd_25185.png emd_25185.png | 49.3 KB | ||
| Filedesc metadata | emd-25185.cif.gz | 5.7 KB | ||
| Filedesc structureFactors | emd_25185_sf.cif.gz | 484.8 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-25185ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25185 http://ftp.pdbj.org/pub/emdb/structures/EMD-25185ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25185 | HTTPS FTP |
-Related structure data
| Related structure data |  7skxMC  7skwC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |

-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_25185.map.gz / Format: CCP4 / Size: 10.3 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | 1.5A resolution 2mFo-dFc MicroED map of proteinase K determined ab initio by electron-counted data | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. generated in cubic-lattice coordinate | ||||||||||||||||||||||||||||||||||||
| Voxel size | X: 0.37267 Å / Y: 0.37267 Å / Z: 0.37076 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 96 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 X (Sec.)
X (Sec.) Y (Row.)
Y (Row.) Z (Col.)
Z (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : Proteinase K
| Entire | Name: Proteinase K |
|---|---|
| Components |
|
-Supramolecule #1: Proteinase K
| Supramolecule | Name: Proteinase K / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Parengyodontium album (fungus) |
| Molecular weight | Theoretical: 28 KDa |
-Macromolecule #1: Proteinase K
| Macromolecule | Name: Proteinase K / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO / EC number: peptidase K |
|---|---|
| Source (natural) | Organism: Parengyodontium album (fungus) |
| Molecular weight | Theoretical: 28.930783 KDa |
| Sequence | String: AAQTNAPWGL ARISSTSPGT STYYYDESAG QGSCVYVIDT GIEASHPEFE GRAQMVKTYY YSSRDGNGHG THCAGTVGSR TYGVAKKTQ LFGVKVLDDN GSGQYSTIIA GMDFVASDKN NRNCPKGVVA SLSLGGGYSS SVNSAAARLQ SSGVMVAVAA G NNNADARN ...String: AAQTNAPWGL ARISSTSPGT STYYYDESAG QGSCVYVIDT GIEASHPEFE GRAQMVKTYY YSSRDGNGHG THCAGTVGSR TYGVAKKTQ LFGVKVLDDN GSGQYSTIIA GMDFVASDKN NRNCPKGVVA SLSLGGGYSS SVNSAAARLQ SSGVMVAVAA G NNNADARN YSPASEPSVC TVGASDRYDR RSSFSNYGSV LDIFGPGTSI LSTWIGGSTR SISGTSMATP HVAGLAAYLM TL GKTTAAS ACRYIADTAN KGDLSNIPFG TVNLLAYNNY QA UniProtKB: Proteinase K |
-Macromolecule #2: 5-amino-2,4,6-triiodobenzene-1,3-dicarboxylic acid
| Macromolecule | Name: 5-amino-2,4,6-triiodobenzene-1,3-dicarboxylic acid / type: ligand / ID: 2 / Number of copies: 2 / Formula: I3C |
|---|---|
| Molecular weight | Theoretical: 558.835 Da |
| Chemical component information | 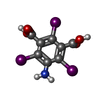 ChemComp-I3C: |
-Macromolecule #3: CALCIUM ION
| Macromolecule | Name: CALCIUM ION / type: ligand / ID: 3 / Number of copies: 5 / Formula: CA |
|---|---|
| Molecular weight | Theoretical: 40.078 Da |
-Macromolecule #4: water
| Macromolecule | Name: water / type: ligand / ID: 4 / Number of copies: 234 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | electron crystallography |
| Aggregation state | 3D array |
-Sample preparation
| Concentration | 5 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY / Support film - Film thickness: 10 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. / Pretreatment - Atmosphere: AIR / Details: NEGATIVE |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 277 K / Instrument: LEICA PLUNGER |
| Details | Milled microcrystals |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 77.0 K / Max: 90.0 K |
| Image recording | Film or detector model: FEI FALCON IV (4k x 4k) / Digitization - Dimensions - Width: 2048 pixel / Digitization - Dimensions - Height: 2048 pixel / Number grids imaged: 1 / Number real images: 1 / Number diffraction images: 840 / Average exposure time: 0.5 sec. / Average electron dose: 0.001 e/Å2 Details: 0.15 degrees per second, 0.5 second readout, 30 to -30 degrees |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: DIFFRACTION / Cs: 2.7 mm / Camera length: 1738 mm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN / Tilt angle: -30.0, 30.0 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Details | Binned by 2. |
|---|---|
| Final reconstruction | Resolution method: DIFFRACTION PATTERN/LAYERLINES / Software - Name: REFMAC |
| Merging software list | Software - Name: AIMLESS |
| Crystallography statistics | Number intensities measured: 416133 / Number structure factors: 39303 / Fourier space coverage: 98.87 / R sym: 0.087 / R merge: 0.277 / Overall phase error: 20 / Overall phase residual: 0 / Phase error rejection criteria: None / High resolution: 1.5 Å Details: Phases were determined by placing 4 ideal helix fragments. These were extended by chain tracing and density modifications. Shell - Shell ID: 1 / Shell - High resolution: 1.53 Å / Shell - Low resolution: 1.5 Å / Shell - Number structure factors: 1758 / Shell - Phase residual: 30 / Shell - Fourier space coverage: 89.3 / Shell - Multiplicity: 8.9 |
-Atomic model buiding 1
| Refinement | Space: RECIPROCAL / Protocol: AB INITIO MODEL / Overall B value: 14.137 / Target criteria: Maximum likelihood |
|---|---|
| Output model | PDB-7skx: |
