 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-16102: Structure of GroEL-nucleotide complex in ADP-like conformation pl... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of GroEL-nucleotide complex in ADP-like conformation plunged 13 ms after mixing with ATP | ||||||||||||
 Map data Map data | |||||||||||||
 Sample Sample |
| ||||||||||||
 Keywords Keywords | GroEL / CHAPERONE | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationGroEL-GroES complex / chaperonin ATPase / virion assembly / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / : / response to heat / protein refolding / protein folding ...GroEL-GroES complex / chaperonin ATPase / virion assembly / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / : / response to heat / protein refolding / protein folding / magnesium ion binding / ATP hydrolysis activity / ATP binding / membrane / identical protein binding / cytosol Similarity search - Function | ||||||||||||
| Biological species |  | ||||||||||||
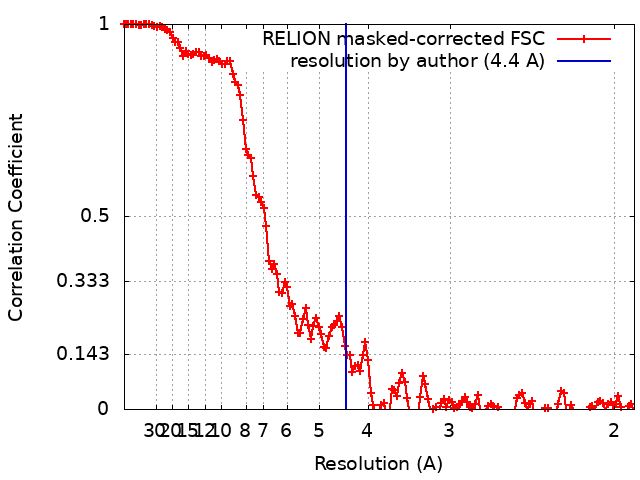
| Method | single particle reconstruction / cryo EM / Resolution: 4.4 Å | ||||||||||||
 Authors Authors | Dhurandhar M / Torino S / Efremov R | ||||||||||||
| Funding support | European Union,  Belgium, 3 items Belgium, 3 items
| ||||||||||||
 Citation Citation | Journal: Nat Methods / Year: 2023 Title: Time-resolved cryo-EM using a combination of droplet microfluidics with on-demand jetting. Authors: Stefania Torino / Mugdha Dhurandhar / Annelore Stroobants / Raf Claessens / Rouslan G Efremov / Abstract: Single-particle cryogenic electron microscopy (cryo-EM) allows reconstruction of high-resolution structures of proteins in different conformations. Protein function often involves transient ...Single-particle cryogenic electron microscopy (cryo-EM) allows reconstruction of high-resolution structures of proteins in different conformations. Protein function often involves transient functional conformations, which can be resolved using time-resolved cryo-EM (trEM). In trEM, reactions are arrested after a defined delay time by rapid vitrification of protein solution on the EM grid. Despite the increasing interest in trEM among the cryo-EM community, making trEM samples with a time resolution below 100 ms remains challenging. Here we report the design and the realization of a time-resolved cryo-plunger that combines a droplet-based microfluidic mixer with a laser-induced generator of microjets that allows rapid reaction initiation and plunge-freezing of cryo-EM grids. Using this approach, a time resolution of 5 ms was achieved and the protein density map was reconstructed to a resolution of 2.1 Å. trEM experiments on GroEL:GroES chaperonin complex resolved the kinetics of the complex formation and visualized putative short-lived conformations of GroEL-ATP complex. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_16102.map.gz | 134.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-16102-v30.xmlemd-16102.xml | 18.9 KB 18.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_16102_fsc.xml | 13.9 KB | Display | FSC data file |
| Images | 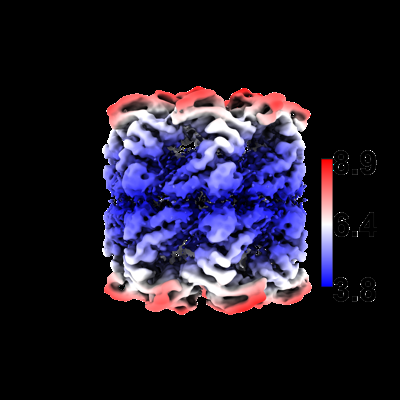 emd_16102.png emd_16102.png | 93.2 KB | ||
| Masks | emd_16102_msk_1.map | 229.8 MB | Mask map | |
| Others | emd_16102_half_map_1.map.gzemd_16102_half_map_2.map.gz | 182.4 MB 182.6 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-16102ftp://ftp.pdbj.org/pub/emdb/structures/EMD-16102 http://ftp.pdbj.org/pub/emdb/structures/EMD-16102ftp://ftp.pdbj.org/pub/emdb/structures/EMD-16102 | HTTPS FTP |
-Related structure data
| Related structure data |  8bl7MC  8bk7C  8bk8C  8bk9C  8bkaC  8bkbC  8bkgC  8bkzC  8bl2C  8blcC  8bldC  8bleC  8blfC  8blyC  8bm0C  8bm1C  8bmdC  8bmoC  8bmtC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_16102.map.gz / Format: CCP4 / Size: 229.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.96 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Mask #1
| File | emd_16102_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
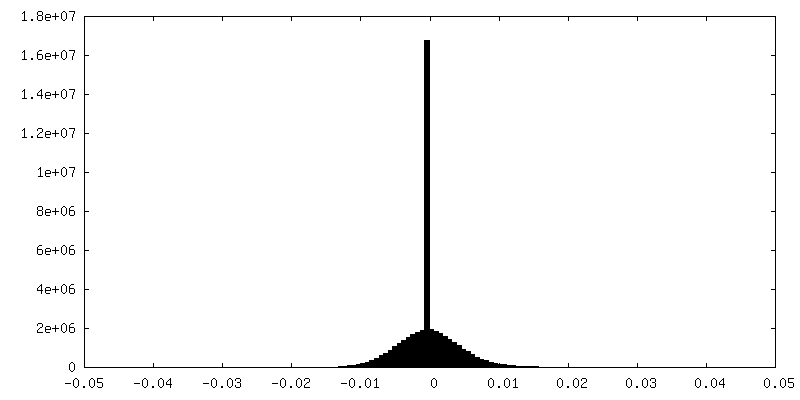
| Density Histograms |






-Half map: #1
| File | emd_16102_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
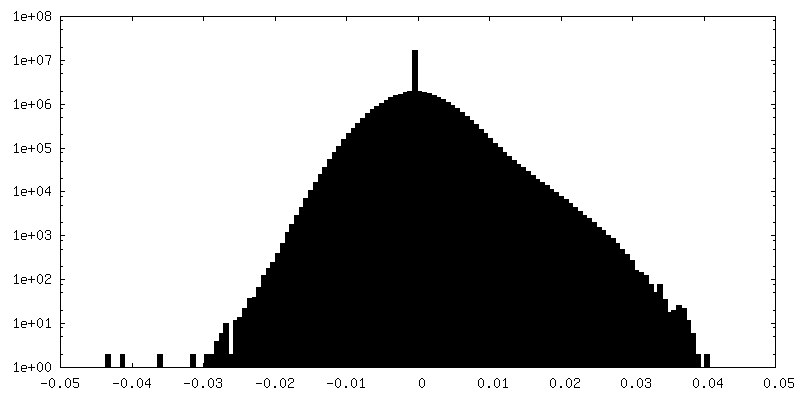
| Density Histograms |






-Half map: #2
| File | emd_16102_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |




- Sample components
Sample components
-Entire : GroEL in complex with ATP
| Entire | Name: GroEL in complex with ATP |
|---|---|
| Components |
|
-Supramolecule #1: GroEL in complex with ATP
| Supramolecule | Name: GroEL in complex with ATP / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 Details: GroEL plunged with 13 ms delay time after mixing with ATP. ADP-like conformation of the 14mer |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 800 KDa |
-Macromolecule #1: Chaperonin GroEL
| Macromolecule | Name: Chaperonin GroEL / type: protein_or_peptide / ID: 1 / Number of copies: 14 / Enantiomer: LEVO / EC number: chaperonin ATPase |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 57.391711 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MAAKDVKFGN DARVKMLRGV NVLADAVKVT LGPKGRNVVL DKSFGAPTIT KDGVSVAREI ELEDKFENMG AQMVKEVASK ANDAAGDGT TTATVLAQAI ITEGLKAVAA GMNPMDLKRG IDKAVTAAVE ELKALSVPCS DSKAIAQVGT ISANSDETVG K LIAEAMDK ...String: MAAKDVKFGN DARVKMLRGV NVLADAVKVT LGPKGRNVVL DKSFGAPTIT KDGVSVAREI ELEDKFENMG AQMVKEVASK ANDAAGDGT TTATVLAQAI ITEGLKAVAA GMNPMDLKRG IDKAVTAAVE ELKALSVPCS DSKAIAQVGT ISANSDETVG K LIAEAMDK VGKEGVITVE DGTGLQDELD VVEGMQFDRG YLSPYFINKP ETGAVELESP FILLADKKIS NIREMLPVLE AV AKAGKPL LIIAEDVEGE ALATLVVNTM RGIVKVAAVK APGFGDRRKA MLQDIATLTG GTVISEEIGM ELEKATLEDL GQA KRVVIN KDTTTIIDGV GEEAAIQGRV AQIRQQIEEA TSDYDREKLQ ERVAKLAGGV AVIKVGAATE VEMKEKKARV EDAL HATRA AVEEGVVAGG GVALIRVASK LADLRGQNED QNVGIKVALR AMEAPLRQIV LNCGEEPSVV ANTVKGGDGN YGYNA ATEE YGNMIDMGIL DPTKVTRSAL QYAASVAGLM ITTECMVTDL PKNDAADLGA AGGMGGMGGM GGMM UniProtKB: Chaperonin GroEL |
-Macromolecule #2: ADENOSINE-5'-TRIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-TRIPHOSPHATE / type: ligand / ID: 2 / Number of copies: 14 / Formula: ATP |
|---|---|
| Molecular weight | Theoretical: 507.181 Da |
| Chemical component information |  ChemComp-ATP: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 8 mg/mL | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| |||||||||||||||||||||
| Grid | Model: Quantifoil R2/1 / Material: COPPER / Support film - #0 - Film type ID: 1 / Support film - #0 - Material: CARBON / Support film - #0 - topology: HOLEY / Support film - #0 - Film thickness: 30 / Support film - #1 - Film type ID: 2 / Support film - #1 - Material: CARBON / Support film - #1 - topology: CONTINUOUS / Support film - #1 - Film thickness: 2 / Pretreatment - Type: PLASMA CLEANING | |||||||||||||||||||||
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | JEOL CRYO ARM 300 |
|---|---|
| Temperature | Min: 100.0 K |
| Specialist optics | Energy filter - Name: In-column Omega Filter |
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Number grids imaged: 15 / Number real images: 2654 / Average electron dose: 63.6 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.55 mm / Nominal defocus max: 3.5 µm / Nominal defocus min: 0.5 µm / Nominal magnification: 60000 |
| Sample stage | Specimen holder model: JEOL CRYOSPECPORTER / Cooling holder cryogen: NITROGEN |
+Image processing
-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT |
|---|---|
| Output model | PDB-8bl7: |
