 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
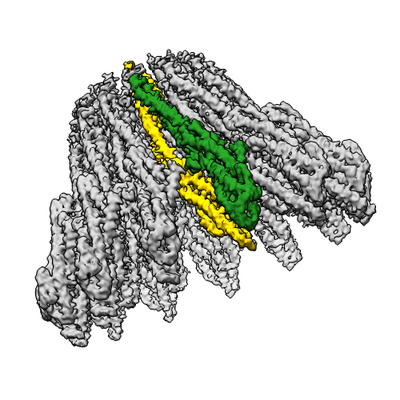
| Title | XaxAB pore complex from Xenorhabdus nematophila | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | bacterial toxin / pore forming-toxins / TOXIN | |||||||||
| Function / homology | : / XaxA / XaxB Function and homology information Function and homology information | |||||||||
| Biological species |  Xenorhabdus nematophila ATCC 19061 (bacteria) Xenorhabdus nematophila ATCC 19061 (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 4.0 Å | |||||||||
 Authors Authors | Schubert E / Vetter IR / Prumbaum D / Penczek PA / Raunser S | |||||||||
| Funding support |  Germany, 1 items Germany, 1 items
| |||||||||
 Citation Citation | Journal: Elife / Year: 2018 Title: Membrane insertion of α-xenorhabdolysin in near-atomic detail. Authors: Evelyn Schubert / Ingrid R Vetter / Daniel Prumbaum / Pawel A Penczek / Stefan Raunser /  Abstract: α-Xenorhabdolysins (Xax) are α-pore-forming toxins (α-PFT) that form 1-1.3 MDa large pore complexes to perforate the host cell membrane. PFTs are used by a variety of bacterial pathogens to attack ...α-Xenorhabdolysins (Xax) are α-pore-forming toxins (α-PFT) that form 1-1.3 MDa large pore complexes to perforate the host cell membrane. PFTs are used by a variety of bacterial pathogens to attack host cells. Due to the lack of structural information, the molecular mechanism of action of Xax toxins is poorly understood. Here, we report the cryo-EM structure of the XaxAB pore complex from and the crystal structures of the soluble monomers of XaxA and XaxB. The structures reveal that XaxA and XaxB are built similarly and appear as heterodimers in the 12-15 subunits containing pore, classifying XaxAB as bi-component α-PFT. Major conformational changes in XaxB, including the swinging out of an amphipathic helix are responsible for membrane insertion. XaxA acts as an activator and stabilizer for XaxB that forms the actual transmembrane pore. Based on our results, we propose a novel structural model for the mechanism of Xax intoxication. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
|---|---|
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_0088.map.gz | 14.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-0088-v30.xmlemd-0088.xml | 13 KB 13 KB | Display Display | EMDB header |
| Images |  emd_0088.png emd_0088.png | 141.2 KB | ||
| Filedesc metadata | emd-0088.cif.gz | 5.6 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-0088ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0088 http://ftp.pdbj.org/pub/emdb/structures/EMD-0088ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0088 | HTTPS FTP |
-Related structure data
| Related structure data |  6gy6MC  6gy7C  6gy8C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |





-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_0088.map.gz / Format: CCP4 / Size: 166.4 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.11 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : XaxAB complex (13 XaxA + 13 XaxB)
| Entire | Name: XaxAB complex (13 XaxA + 13 XaxB) |
|---|---|
| Components |
|
-Supramolecule #1: XaxAB complex (13 XaxA + 13 XaxB)
| Supramolecule | Name: XaxAB complex (13 XaxA + 13 XaxB) / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Xenorhabdus nematophila ATCC 19061 (bacteria) |
-Supramolecule #2: XaxA protomer
| Supramolecule | Name: XaxA protomer / type: complex / ID: 2 / Parent: 1 / Macromolecule list: #1 / Details: 13 XaxA protomers in the XaxAB pore complex |
|---|---|
| Source (natural) | Organism: Xenorhabdus nematophila ATCC 19061 (bacteria) |
-Supramolecule #3: XaxB protomer
| Supramolecule | Name: XaxB protomer / type: complex / ID: 3 / Parent: 1 / Macromolecule list: #2 / Details: 13 XaxB protomers in the XaxAB pore complex |
|---|---|
| Source (natural) | Organism: Xenorhabdus nematophila ATCC 19061 (bacteria) |
-Macromolecule #1: XaxA
| Macromolecule | Name: XaxA / type: protein_or_peptide / ID: 1 / Number of copies: 13 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Xenorhabdus nematophila ATCC 19061 (bacteria) |
| Molecular weight | Theoretical: 47.465859 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MENDMSSNQT LAEKKIPVSE VPSATLKMLT SQAEGVARPG GIFTKGDLIN IKLYVKHSLE LPFTLEGVKE YIGYNDIDID GLKPAKMAT LFKEIHDHAL SWSGVESKVQ QQSIDLENAG KQITLTGDEI ISVIDQMPII ERVKNKLGDL TDKQLAEITY T NDDKEIAV ...String: MENDMSSNQT LAEKKIPVSE VPSATLKMLT SQAEGVARPG GIFTKGDLIN IKLYVKHSLE LPFTLEGVKE YIGYNDIDID GLKPAKMAT LFKEIHDHAL SWSGVESKVQ QQSIDLENAG KQITLTGDEI ISVIDQMPII ERVKNKLGDL TDKQLAEITY T NDDKEIAV ELGNILESMK KDIKRQQENT QKVKTAVSDF KLKLIGGELS DGTIAQGLQP QISSKKKLMD DNNLSTTIKD LQ SKIDEKN KEIDQFQKDY NKYVGLAFSG MVGGIISWAI TGGIFGDKAE KARKQKNKLI DEVKDLQSQV KDKSALQTSV QNL SLSFAG IHTSMVDAEE ALNHLDFMWN TMLTQITTSR DKFDDINDAL KLTSFVIAFK QVIEPWRDVQ GSAAQLIQTF DEAL AEYKK LYHGTLEVLF QGPHHHHHH UniProtKB: XaxA |
-Macromolecule #2: XaxB
| Macromolecule | Name: XaxB / type: protein_or_peptide / ID: 2 / Number of copies: 13 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Xenorhabdus nematophila ATCC 19061 (bacteria) |
| Molecular weight | Theoretical: 38.518688 KDa |
| Recombinant expression | Organism: |
| Sequence | String: YPEINIKAMN QAVNTIWLLA QRQTSGIEII NDKVKRISAY SREFDEMMRD SLAQLAPVLK QLTSDAAFQT IAQIDEALAD PSLSKDDRE ALTLERNNLI QNLSKHIDNV IVSFTGRTSK LTNKISDISD MVIAERLQDL VTQTESQKTE LQSDIDPKTE K RNKLDADR ...String: YPEINIKAMN QAVNTIWLLA QRQTSGIEII NDKVKRISAY SREFDEMMRD SLAQLAPVLK QLTSDAAFQT IAQIDEALAD PSLSKDDRE ALTLERNNLI QNLSKHIDNV IVSFTGRTSK LTNKISDISD MVIAERLQDL VTQTESQKTE LQSDIDPKTE K RNKLDADR EKIIESQDVI RQNNIADMFK DFIPSAKDID GLDFTQPKKE AIKQAIKQGA EIARKILGKV SEGLKYIDLA DA RMKLSDQ IDQLITETDE LKAKIREVEL RLSGLKDVMQ IDTERTTLLT EAVKIEQVWI SFAEQLHKLS NDEINQQDLS NLI NGQLDF LNNLTLQYNK LK UniProtKB: XaxB |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Grid | Model: C-flat-2/1 |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 90 % / Chamber temperature: 298.15 K / Instrument: GATAN CRYOPLUNGE 3 |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: COUNTING / Average electron dose: 44.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: SPOT SCAN / Imaging mode: BRIGHT FIELD |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
