 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6eaj: CRYSTAL STRUCTURE OF HUMAN RESPIRATORY SYNCYTIAL VIRUS FUSION GLY... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6eaj | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF HUMAN RESPIRATORY SYNCYTIAL VIRUS FUSION GLYCOPROTEIN INHIBITOR ESCAPE VARIANT T400A STABILIZED IN THE PREFUSION STATE | |||||||||
 Components Components | Fusion glycoprotein F0 | |||||||||
 Keywords Keywords |  VIRAL PROTEIN / CLASS I VIRAL FUSION PROTEIN / FUSION / RESPIRATORY SYNCYTIAL VIRUS / PREFUSION / FUSION INHIBITOR VIRAL PROTEIN / CLASS I VIRAL FUSION PROTEIN / FUSION / RESPIRATORY SYNCYTIAL VIRUS / PREFUSION / FUSION INHIBITOR | |||||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of syncytium formation by virus / host cell Golgi membrane / entry receptor-mediated virion attachment to host cell / symbiont entry into host cell / fusion of virus membrane with host plasma membrane / viral envelope / host cell plasma membrane / virion membrane / membrane / identical protein binding / plasma membraneSimilarity search - Function | |||||||||
| Biological species |  Human respiratory syncytial virus Human respiratory syncytial virus | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.851 Å | |||||||||
 Authors Authors | Battles, M.B. / McLellan, J.S. | |||||||||
| Funding support |  United States, 2items United States, 2items
| |||||||||
 Citation Citation | Journal: To be published Title: Structural Basis for Respiratory Syncytial Virus Fusion Inhibitor Resistance Authors: Battles, M.B. / McLellan, J.S. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6eaj.cif.gz | 106.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6eaj.ent.gz | 78.2 KB | Display | PDB format |
| PDBx/mmJSON format | 6eaj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ea/6eajftp://data.pdbj.org/pub/pdb/validation_reports/ea/6eaj | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6eadC  6eaeC  6eafC  6eagC  6eahC  6eaiC  6eakC  6ealC  6eamC  6eanC  5ea4S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 63188.320 Da / Num. of mol.: 1 / Fragment: RSV F ectodomain / Mutation: S155C, S290C, S190F, V207L, T400A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Human respiratory syncytial virus / Plasmid: P(ALPHA)H / Production host:  Homo sapiens (human) / Strain (production host): HEK293 FREESTYLE / References: UniProt: W8RJF9, UniProt: P03420*PLUS Homo sapiens (human) / Strain (production host): HEK293 FREESTYLE / References: UniProt: W8RJF9, UniProt: P03420*PLUS | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-NHE / | CHES (buffer)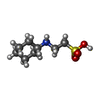  Mass: 207.290 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H17NO3S / Comment: pH buffer*YM Mass: 207.290 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H17NO3S / Comment: pH buffer*YM#4: Chemical | ChemComp-TAR / | Tartaric acid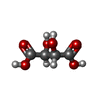  Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 12 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 12 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.14 Å3/Da / Density % sol: 60.79 % / Mosaicity: 0.16 ° |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 9.5 / Details: 1.50M K/Na tartrate, 0.2M LiSO4, 0.1M CHES pH 9.5 |
-Data collection
| Diffraction | Mean temperature: 80 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL / Beamline: BL14-1 / Wavelength: 1.181 Å |
| Detector | Type: MARMOSAIC 325 mm CCD / Detector: CCD / Date: Jun 15, 2016 |
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.181 Å / Relative weight: 1 |
| Reflection | Resolution: 2.85→56.06 Å / Num. obs: 19529 / % possible obs: 99.7 % / Redundancy: 4.8 % / Biso Wilson estimate: 60.99 Å2 / CC1/2: 0.993 / Rmerge(I) obs: 0.141 / Rpim(I) all: 0.072 / Rrim(I) all: 0.159 / Net I/σ(I): 8.6 / Num. measured all: 92996 / Scaling rejects: 18 |
| Reflection shell | Resolution: 2.85→3 Å / Redundancy: 4.9 % / Rmerge(I) obs: 0.974 / Num. unique obs: 2775 / CC1/2: 0.539 / Rpim(I) all: 0.489 / Rrim(I) all: 1.094 / % possible all: 99.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5EA4 Resolution: 2.851→56.06 Å / SU ML: 0.4 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 23.75
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 173.91 Å2 / Biso mean: 60.1719 Å2 / Biso min: 24.49 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.851→56.06 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 7
|
