 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4rmp: Crystal structure of allophycocyanin from marine cyanobacterium P... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4rmp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of allophycocyanin from marine cyanobacterium Phormidium sp. A09DM | ||||||
 Components Components | (Allophycocyanin ) x 2 ) x 2 | ||||||
 Keywords Keywords | PHOTOSYNTHESIS / Phycocyanobilin chromophore / Globin-like fold (SCOP / 46457) / Light harvesting Phycobiliprotein | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Phormidium rubidum A09DM (bacteria) Phormidium rubidum A09DM (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.506 Å | ||||||
 Authors Authors | Kumar, V. / Gupta, G.D. / Sonani, R.R. / Madamwar, D. | ||||||
 Citation Citation | Journal: Plos One / Year: 2015 Title: Crystal Structure of Allophycocyanin from Marine Cyanobacterium Phormidium sp. A09DM. Authors: Sonani, R.R. / Gupta, G.D. / Madamwar, D. / Kumar, V. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4rmp.cif.gz | 80.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4rmp.ent.gz | 60.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4rmp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rm/4rmpftp://data.pdbj.org/pub/pdb/validation_reports/rm/4rmp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1kn1S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | THE ASYMMETRIC UNIT CONTAINS ONE EACH A AND B-CHAINS WHICH FORM A HETERODIMER (KNOWN AS ALPHA/BETA MONOMER). THREE ALPHA/BETA MONOMERS FORM A BIOLOGICAL UNIT. THE HEXAMER (TRIMER of ALPHA/BETA MONOMERS) IS GENERATED BY CRYSTALLOGRAPHIC SYMMETRY |
-Components
| #1: Protein | Mass: 17492.889 Da / Num. of mol.: 1 / Source method: isolated from a natural source Details: Marine cyanobacterium isolated from rocky shores of Gujarat Source: (natural) Phormidium rubidum A09DM (bacteria) / References: UniProt: A0A078K1U6 | ||||
|---|---|---|---|---|---|
| #2: Protein | Mass: 17438.807 Da / Num. of mol.: 1 / Source method: isolated from a natural source Details: Marine cyanobacterium isolated from rocky shores of Gujarat Source: (natural) Phormidium rubidum A09DM (bacteria) / References: UniProt: A0A078K4M9 | ||||
| #3: Chemical | Phycocyanobilin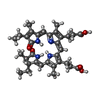  Mass: 588.694 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C33H40N4O6 Mass: 588.694 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C33H40N4O6#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 179 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 179 / Source method: isolated from a natural source / Formula: H2OSequence details | NATURAL VARIANTS OF THE UNP REFERENCE, AS CONFIRMED BY NUCLEOTIDE | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.73 Å3/Da / Density % sol: 54.87 % |
|---|---|
| Crystal grow | Temperature: 288 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: 20% (w/v) PEG 6000, 0.1M bicine, pH 8.5, VAPOR DIFFUSION, SITTING DROP, temperature 288K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: BRUKER AXS MICROSTAR / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jun 17, 2014 / Details: HELIOS Cu X-RAY OPTICS |
| Radiation | Monochromator: HELIOS Cu X-RAY OPTICS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.506→35 Å / Num. all: 13362 / Num. obs: 13362 / % possible obs: 99.9 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 10.8 % / Biso Wilson estimate: 41.97 Å2 / Rmerge(I) obs: 0.109 / Net I/σ(I): 22.8 |
| Reflection shell | Resolution: 2.506→2.64 Å / Redundancy: 10.7 % / Rmerge(I) obs: 0.557 / Mean I/σ(I) obs: 5.3 / Num. unique all: 1903 / % possible all: 99.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1KN1 Resolution: 2.506→29.226 Å / SU ML: 0.28 / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 24.82 / Stereochemistry target values: ML Details: The covalent bonds between S atoms of Cys-81 and the chromophore (CYC) were restrained to a distance of 1.8A for both A and B chains
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 36.17 Å2 | ||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.266 Å | ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.506→29.226 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / % reflection obs: 100 %
|
