 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3zci: Crystal structure of Helicobacter pylori T4SS protein CagL in a c... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3zci | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Helicobacter pylori T4SS protein CagL in a cubic crystal form with a distorted helical conformation of the RGD-motif | ||||||
 Components Components | CAG PATHOGENICITY ISLAND PROTEIN (CAG18) | ||||||
 Keywords Keywords |  PROTEIN BINDING / KKQEK / SER MUTANT / ADHESION / RGD MOTIF / INTEGRIN BINDING / TYPE IV SECRETION / T4S / VIRULENCE / CAG18 / HP0539 PROTEIN BINDING / KKQEK / SER MUTANT / ADHESION / RGD MOTIF / INTEGRIN BINDING / TYPE IV SECRETION / T4S / VIRULENCE / CAG18 / HP0539 | ||||||
| Function / homology | Methane Monooxygenase Hydroxylase; Chain G, domain 1 - #1660 / Methane Monooxygenase Hydroxylase; Chain G, domain 1 / Up-down Bundle / Mainly Alpha / Meso-2,3-Butanediol / 1,4-DIETHYLENE DIOXIDE / Cag pathogenicity island protein (Cag18) Function and homology information Function and homology information | ||||||
| Biological species |   HELICOBACTER PYLORI (bacteria) HELICOBACTER PYLORI (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.201 Å | ||||||
 Authors Authors | Barden, S. / Niemann, H.H. | ||||||
 Citation Citation | Journal: Structure / Year: 2013 Title: A Helical Rgd Motif Promoting Cell Adhesion: Crystal Structures of the Helicobacter Pylori Type Iv Secretion System Pilus Protein Cagl Authors: Barden, S. / Lange, S. / Tegtmeyer, N. / Conradi, J. / Sewald, N. / Backert, S. / Niemann, H.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3zci.cif.gz | 59.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3zci.ent.gz | 49.3 KB | Display | PDB format |
| PDBx/mmJSON format | 3zci.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zc/3zciftp://data.pdbj.org/pub/pdb/validation_reports/zc/3zci | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 24940.545 Da / Num. of mol.: 1 / Fragment: RESIDUES 21-237 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) HELICOBACTER PYLORI (bacteria) / Strain: 26695 / Production host: ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / Variant (production host): CODONPLUS RIL / References: UniProt: O25272 |
|---|
-Non-polymers , 5 types, 123 molecules 
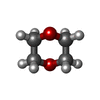
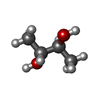


| #2: Chemical | Sulfate Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#3: Chemical | 1,4-Dioxane Mass: 88.105 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H8O2 Mass: 88.105 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H8O2#4: Chemical |  Mass: 90.121 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H10O2 Mass: 90.121 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H10O2#5: Chemical | ChemComp-CL / | Chloride Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 113 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Nonpolymer details | MESO-2,3-BUTANEDIOL (BU9): CRYO PROTECTANT 1,4-DIETHYLENE DIOXIDE (DIO): CRYSTALLIZATION COCKTAIL ...MESO-2,3-BUTANEDIOL |
|---|---|
| Sequence details | SURFACE ENTROPY REDUCTION MUTANT KKQEK |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 3 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5 Å3/Da / Density % sol: 75 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion / pH: 6.5 Details: PROTEIN CONCENTRATION:7 MG/ML. RESERVOIR: 100 MM MES PH6.5, 1.2 M AMMONIUM SULPHATE, 2-4 % 1,4 DIOXANE. VAPOR DIFFUSION AT 277 K WITH DROP RATIO OF |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.2 / Wavelength: 0.97814, 0.97814, 0.97847 / Beamline: 14.2 / Wavelength: 0.97814, 0.97814, 0.97847 | |||||||||
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Mar 2, 2011 / Details: MIRRORS | |||||||||
| Radiation | Monochromator: DOUBLE CRYSTAL SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 2.2→20 Å / Num. obs: 24942 / % possible obs: 99.9 % / Observed criterion σ(I): -3 / Redundancy: 14.4 % / Biso Wilson estimate: 37.06 Å2 / Rmerge(I) obs: 0.14 / Net I/σ(I): 12.7 | |||||||||
| Reflection shell | Resolution: 2.2→2.32 Å / Redundancy: 9.2 % / Rmerge(I) obs: 0.91 / Mean I/σ(I) obs: 2.3 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD Starting model: NONE Resolution: 2.201→19.868 Å / SU ML: 0.36 / σ(F): 1.33 / Phase error: 23.4 / Stereochemistry target values: ML / Details: SECONDARY STRUCTURE RESTRAINTS APPLIED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 59.58 Å2 / ksol: 0.314 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 46.39 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.201→19.868 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
