 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3m9k: Crystal structure of human thioredoxin C69/73S double-mutant, oxi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3m9k | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human thioredoxin C69/73S double-mutant, oxidized form | ||||||
 Components Components | Thioredoxin | ||||||
 Keywords Keywords | OXIDOREDUCTASE / dimer / intermolecular disulfide bond / DTT / disulfide bond / S-nitrosylation | ||||||
| Function / homology |  Function and homology information Function and homology informationProtein repair / cellular detoxification of hydrogen peroxide / positive regulation of peptidyl-cysteine S-nitrosylation / protein-disulfide reductase (NAD(P)H) activity / thioredoxin-disulfide reductase (NADPH) activity / Interconversion of nucleotide di- and triphosphates / negative regulation of protein export from nucleus / Regulation of FOXO transcriptional activity by acetylation / NFE2L2 regulating anti-oxidant/detoxification enzymes / response to nitric oxide ...Protein repair / cellular detoxification of hydrogen peroxide / positive regulation of peptidyl-cysteine S-nitrosylation / protein-disulfide reductase (NAD(P)H) activity / thioredoxin-disulfide reductase (NADPH) activity / Interconversion of nucleotide di- and triphosphates / negative regulation of protein export from nucleus / Regulation of FOXO transcriptional activity by acetylation / NFE2L2 regulating anti-oxidant/detoxification enzymes / response to nitric oxide / Detoxification of Reactive Oxygen Species / The NLRP3 inflammasome / protein-disulfide reductase activity / positive regulation of DNA binding / Purinergic signaling in leishmaniasis infection / activation of protein kinase B activity / cell redox homeostasis / TP53 Regulates Metabolic Genes / response to radiation / positive regulation of peptidyl-serine phosphorylation / Oxidative Stress Induced Senescence / positive regulation of phosphatidylinositol 3-kinase/protein kinase B signal transduction / negative regulation of transcription by RNA polymerase II / protein homodimerization activity / RNA binding / extracellular exosome / extracellular region / nucleoplasm / nucleus / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | ||||||
 Authors Authors | Weichsel, A. / Montfort, W.R. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2010 Title: Crystal structure of human thioredoxin revealing an unraveled helix and exposed S-nitrosation site. Authors: Weichsel, A. / Kem, M. / Montfort, W.R. #1: Journal: Biochemistry / Year: 2007Title: Buried S-nitrosocysteine revealed in crystal structures of human thioredoxin. Authors: Weichsel, A. / Brailey, J.L. / Montfort, W.R. #2: Journal: Structure / Year: 1996Title: Crystal structures of reduced, oxidized, and mutated human thioredoxins: evidence for a regulatory homodimer. Authors: Weichsel, A. / Gasdaska, J.R. / Powis, G. / Montfort, W.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3m9k.cif.gz | 109.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3m9k.ent.gz | 85.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3m9k.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/m9/3m9kftp://data.pdbj.org/pub/pdb/validation_reports/m9/3m9k | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3m9jC  1ertS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj














- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 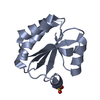
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / Trx / ATL-derived factor / ADF / Surface-associated sulphydryl protein / SASP Mass: 11718.348 Da / Num. of mol.: 2 / Mutation: C69S, C73S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: TXN, TRDX, TRX, TRX1 / Plasmid: pET3a / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3) pLysS / References: UniProt: P10599 Escherichia coli (E. coli) / Strain (production host): BL21(DE3) pLysS / References: UniProt: P10599#2: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-D1D / ( | 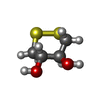  Mass: 152.235 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H8O2S2 Mass: 152.235 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H8O2S2#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 165 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 165 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.26 Å3/Da / Density % sol: 62.28 % |
|---|---|
| Crystal grow | Temperature: 298 K / pH: 5.6 Details: 1.8 M ammonium sulfate, 100 mM MES, 10 mM CoCl2, 2 mM DTT, pH 5.6, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-2 / Wavelength: 0.97946 / Beamline: BL9-2 / Wavelength: 0.97946 |
| Detector | Type: MARMOSAIC 325 mm CCD / Detector: CCD / Date: Apr 17, 2009 / Details: RH COATED FLAT MIRROR, TOROIDAL FOCUSING MIRROR |
| Radiation | Monochromator: SI(111) DOUBLE CRYSTAL MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97946 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→22.43 Å / Num. obs: 48316 / % possible obs: 98.6 % / Observed criterion σ(I): 0 / Redundancy: 9.89 % / Biso Wilson estimate: 29.9 Å2 / Rmerge(I) obs: 0.053 / Net I/σ(I): 15.2 |
| Reflection shell | Resolution: 1.5→1.55 Å / Redundancy: 9.16 % / Rmerge(I) obs: 0.631 / Mean I/σ(I) obs: 2.8 / % possible all: 97.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1ERT Resolution: 1.5→22.34 Å / Cor.coef. Fo:Fc: 0.967 / Cor.coef. Fo:Fc free: 0.958 / Occupancy max: 1 / Occupancy min: 0 / SU B: 2.812 / SU ML: 0.047 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.077 / ESU R Free: 0.069 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES: REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.87 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.202 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→22.34 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.5→1.54 Å / Total num. of bins used: 20
|
