Entry Database : PDB / ID : 3llmTitle Crystal Structure Analysis of a RNA Helicase ATP-dependent RNA helicase A Keywords / / / / / / / / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 2.8 Å Authors Schutz, P. / Karlberg, T. / Collins, R. / Arrowsmith, C.H. / Berglund, H. / Bountra, C. / Flodin, S. / Flores, A. / Graslund, S. / Hammarstrom, M. ...Schutz, P. / Karlberg, T. / Collins, R. / Arrowsmith, C.H. / Berglund, H. / Bountra, C. / Flodin, S. / Flores, A. / Graslund, S. / Hammarstrom, M. / Johansson, A. / Johansson, I. / Kallas, A. / Kraulis, P. / Kotenyova, T. / Kotzsch, A. / Markova, N. / Moche, M. / Nielsen, T.K. / Nordlund, P. / Nyman, T. / Persson, C. / Roos, A.K. / Siponen, M.I. / Svensson, L. / Thorsell, A.G. / Tresaugues, L. / Van Den Berg, S. / Wahlberg, E. / Weigelt, J. / Welin, M. / Wisniewska, M. / Schuler, H.M. / Structural Genomics Consortium (SGC) Journal : J.Mol.Biol. / Year : 2010Title : Crystal structure of human RNA helicase A (DHX9): structural basis for unselective nucleotide base binding in a DEAD-box variant protein.Authors : Schutz, P. / Wahlberg, E. / Karlberg, T. / Hammarstrom, M. / Collins, R. / Flores, A. / Schuler, H. History Deposition Jan 29, 2010 Deposition site / Processing site Revision 1.0 May 12, 2010 Provider / Type Revision 1.1 Jul 13, 2011 Group
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords HYDROLASE / alpha-beta-alpha /
HYDROLASE / alpha-beta-alpha /  Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj







 Assembly
Assembly





 Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn
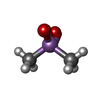
Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 136.989 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6AsO2
Mass: 136.989 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6AsO2 Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM
Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Sample preparation
Sample preparation / Beamline: I03 / Wavelength: 0.98000, 0.98020, 0.96860
/ Beamline: I03 / Wavelength: 0.98000, 0.98020, 0.96860 Processing
Processing