 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2oju: X-ray structure of complex of human cyclophilin J with cyclosporin A -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2oju | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray structure of complex of human cyclophilin J with cyclosporin A | ||||||
 Components Components |
| ||||||
 Keywords Keywords | ISOMERASE/IMMUNOSUPPRESSANT / ISOMERASE-IMMUNOSUPPRESSANT COMPLEX / CYCLOPHILIN-CYCLOSPORIN COMPLEX /  CYCLOSPORIN A / IMMUNOSUPPRESSANT / CYCLOPHILIN CYCLOSPORIN A / IMMUNOSUPPRESSANT / CYCLOPHILIN | ||||||
| Function / homology |  Function and homology information Function and homology informationcatalytic step 2 spliceosome / mRNA Splicing - Major Pathway / peptidylprolyl isomerase / peptidyl-prolyl cis-trans isomerase activity / mRNA splicing, via spliceosome / protein folding / nucleoplasmSimilarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) TOLYPOCLADIUM INFLATUM (fungus) TOLYPOCLADIUM INFLATUM (fungus) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Xia, Z. / Huang, L. | ||||||
 Citation Citation | Journal: To be Published Title: Targeting Cyclophilin J, a Novel Peptidyl-Prolyl Isomerase, Can Induce Cellular G1/S Arrest and Repress the Growth of Hepatocellular Carcinoma Authors: Chen, J. / Chen, S. / Huang, L. / Zhao, X. / Tan, J. / Huang, C. / Saiyin, H. / Zhang, M. / Zeng, X. / Xi, J. / Wan, B. / Zhao, Y. / Xia, Z. / Jiang, H. / Yi, Q. / Liu, J.O. / Yu, L. | ||||||
| History |
| ||||||
| Remark 650 | HELIX DETERMINATION METHOD: AUTHOR DETERMINED |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2oju.cif.gz | 84 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2oju.ent.gz | 63.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2oju.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/oj/2ojuftp://data.pdbj.org/pub/pdb/validation_reports/oj/2oju | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2ok3S S: Starting model for refinement |
|---|---|
| Similar structure data |





































-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 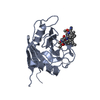
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18750.143 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Tissue: FETAL BRAIN / Gene: PPIL3B / Plasmid: PTXB1 / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): ER2566 / References: UniProt: Q9H2H8, peptidylprolyl isomerase ESCHERICHIA COLI (E. coli) / Strain (production host): ER2566 / References: UniProt: Q9H2H8, peptidylprolyl isomerase#2: Protein/peptide | Ciclosporin / CICLOSPORIN / CICLOSPORINE / Ciclosporin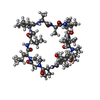  Type: Cyclic peptide / Class: ImmunosuppressantImmunosuppressive drug / Mass: 1220.625 Da / Num. of mol.: 2 / Source method: obtained synthetically Type: Cyclic peptide / Class: ImmunosuppressantImmunosuppressive drug / Mass: 1220.625 Da / Num. of mol.: 2 / Source method: obtained syntheticallyDetails: CYCLOSPORIN IS A CYCLIC UNDECAPEPTIDE. CYCLIZATION IS ACHIEVED BY LINKING THE N- AND THE C- TERMINI. Source: (synth.) TOLYPOCLADIUM INFLATUM (fungus) / References: NOR: NOR00033, Cyclosporin A#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 32 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 32 / Source method: isolated from a natural source / Formula: H2OCompound details | CYCLOSPORI | Sequence details | THE N-TERMINAL 6 RESIDUES 'SRVDGG' IN CHAINS A AND B IS AN ENGINEERED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.04 Å3/Da / Density % sol: 59.63 % |
|---|---|
| Crystal grow | pH: 7.4 Details: 0.1MOL/L TRIS-HCL(PH=7.4), 6%(V/V) DMSO, 18%(W/V) PEG8000, VAPOR DIFFUSION, HANGING DROP, TEMPERATURE 293.0K |
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BSRF  / Beamline: 3W1A / Wavelength: 0.9826 / Beamline: 3W1A / Wavelength: 0.9826 |
| Detector | Detector: CCD / Date: Feb 26, 2005 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9826 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→50 Å / Num. obs: 19272 / % possible obs: 97.6 % / Observed criterion σ(I): 0 / Biso Wilson estimate: 42.5 Å2 / Rmerge(I) obs: 0.076 |
| Reflection shell | Resolution: 2.4→2.49 Å / Rmerge(I) obs: 0.406 / % possible all: 83.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2OK3 Resolution: 2.4→30 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 220039.63 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 33.27 Å2 / ksol: 0.28 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 51.3 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.49 Å / Rfactor Rfree error: 0.037 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
