 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2il4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of At1g77540-Coenzyme A Complex | |||||||||
 Components Components | Protein At1g77540 | |||||||||
 Keywords Keywords |  TRANSFERASE / CoA / Coenzyme-A / COG2388 Family / acetyltransferase / At1g77540 / STRUCTURAL GENOMICS FUNCTIONAL FOLLOW-UP STUDY / PROTEIN STRUCTURE INITIATIVE / PSI / CENTER FOR EUKARYOTIC STRUCTURAL GENOMICS / CESG TRANSFERASE / CoA / Coenzyme-A / COG2388 Family / acetyltransferase / At1g77540 / STRUCTURAL GENOMICS FUNCTIONAL FOLLOW-UP STUDY / PROTEIN STRUCTURE INITIATIVE / PSI / CENTER FOR EUKARYOTIC STRUCTURAL GENOMICS / CESG | |||||||||
| Function / homology |  Function and homology informationhistone acetyltransferase activity / Transferases; Acyltransferases; Transferring groups other than aminoacyl groups / peroxisome Function and homology informationhistone acetyltransferase activity / Transferases; Acyltransferases; Transferring groups other than aminoacyl groups / peroxisomeSimilarity search - Function | |||||||||
| Biological species |  Arabidopsis thaliana (thale cress) Arabidopsis thaliana (thale cress) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.054 Å | |||||||||
 Authors Authors | Bitto, E. / Wesenberg, G.E. / Phillips Jr., G.N. / Bingman, C.A. / Center for Eukaryotic Structural Genomics (CESG) | |||||||||
 Citation Citation | Journal: Biochemistry / Year: 2006 Title: Structure of Arabidopsis thaliana At1g77540 Protein, a Minimal Acetyltransferase from the COG2388 Family. Authors: Tyler, R.C. / Bitto, E. / Berndsen, C.E. / Bingman, C.A. / Singh, S. / Lee, M.S. / Wesenberg, G.E. / Denu, J.M. / Phillips, G.N. / Markley, J.L. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2il4.cif.gz | 36 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2il4.ent.gz | 23 KB | Display | PDB format |
| PDBx/mmJSON format | 2il4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/il/2il4ftp://data.pdbj.org/pub/pdb/validation_reports/il/2il4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1xmtSC  2evnC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |







-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 11752.433 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Arabidopsis thaliana (thale cress) / Gene: At1g77540 / Plasmid: PVP13-GW / Production host:  Escherichia coli (E. coli) / Strain (production host): BL834(DE3) PLACI+RARE / References: UniProt: Q9CAQ2 Escherichia coli (E. coli) / Strain (production host): BL834(DE3) PLACI+RARE / References: UniProt: Q9CAQ2 |
|---|---|
| #2: Chemical | ChemComp-COA / Coenzyme A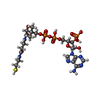  Mass: 767.534 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H36N7O16P3S Mass: 767.534 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H36N7O16P3S |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 61 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 61 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.24 Å3/Da / Density % sol: 45.08 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop Details: PROTEIN SOLUTION (10 MG/ML PROTEIN, 0.050 M SODIUM CHLORIDE, 0.003 M SODIUM AZIDE, 0.0003 M TCEP, 0.005 M MES PH 6.0) MIXED IN A 1:1 RATIO WITH THE WELL SOLUTION (29% PEG 5000, 0.10 M SODIUM ...Details: PROTEIN SOLUTION (10 MG/ML PROTEIN, 0.050 M SODIUM CHLORIDE, 0.003 M SODIUM AZIDE, 0.0003 M TCEP, 0.005 M MES PH 6.0) MIXED IN A 1:1 RATIO WITH THE WELL SOLUTION (29% PEG 5000, 0.10 M SODIUM CITRATE, 0.10 M PIPES PH 6.5). CRYSTALS SOAKED 16 HOURS IN WELL SOLUTION SUPPLEMENTED WITH 0.009 M acetyl-Coenzyme. Crystals cryo-protected with fomblin 2500, vapor diffusion, hanging drop, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 8-BM / Wavelength: 0.97949 Å / Beamline: 8-BM / Wavelength: 0.97949 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Nov 30, 2005 Details: Double crystal monochromator, Si111, 2nd crystal sagital focus followed by Rh coated bent cylinder vertically focusing mirror |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97949 Å / Relative weight: 1 |
| Reflection | Resolution: 2.05→31.98 Å / Num. obs: 6473 / % possible obs: 99.2 % / Redundancy: 4.3 % / Rmerge(I) obs: 0.052 / Χ2: 0.993 / Net I/σ(I): 18.303 |
| Reflection shell | Resolution: 2.05→2.1 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.251 / Mean I/σ(I) obs: 4.993 / Num. unique all: 432 / Χ2: 0.902 / % possible all: 98 |
-Phasing
| Phasing MR | Rfactor: 0.462 / Cor.coef. Fo:Fc: 0.533
|
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1XMT Resolution: 2.054→31.976 Å / Cor.coef. Fo:Fc: 0.966 / Cor.coef. Fo:Fc free: 0.939 / WRfactor Rfree: 0.213 / WRfactor Rwork: 0.16 / SU B: 8.596 / SU ML: 0.125 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.202 / ESU R Free: 0.179 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL PLUS MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 33.963 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.054→31.976 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.054→2.107 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 1.8965 Å / Origin y: 0.2197 Å / Origin z: 3.6025 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection: ALL |
