 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
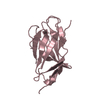
| Entry | Database: PDB / ID: 2bk8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | M1 domain from titin | ||||||
 Components Components | TITIN HEART ISOFORM N2-B | ||||||
 Keywords Keywords |  IG DOMAIN / TITIN / M-BAND / STRUCTURAL PROTEIN / MUSCLE / ANTIBODY IG DOMAIN / TITIN / M-BAND / STRUCTURAL PROTEIN / MUSCLE / ANTIBODY | ||||||
| Function / homology |  Function and homology information Function and homology informationsarcomerogenesis / structural molecule activity conferring elasticity / telethonin binding / skeletal muscle myosin thick filament assembly / cardiac myofibril assembly / muscle alpha-actinin binding / detection of muscle stretch / cardiac muscle tissue morphogenesis / regulation of catalytic activity / cardiac muscle hypertrophy ...sarcomerogenesis / structural molecule activity conferring elasticity / telethonin binding / skeletal muscle myosin thick filament assembly / cardiac myofibril assembly / muscle alpha-actinin binding / detection of muscle stretch / cardiac muscle tissue morphogenesis / regulation of catalytic activity / cardiac muscle hypertrophy / mitotic chromosome condensation / Striated Muscle Contraction / M band / actinin binding / I band / cardiac muscle cell development / regulation of protein kinase activity / sarcomere organization / structural constituent of muscle / skeletal muscle thin filament assembly / striated muscle thin filament / striated muscle contraction / protein kinase A signaling / cardiac muscle contraction / muscle contraction / condensed nuclear chromosome / positive regulation of protein secretion / Z disc / response to calcium ion / : / actin filament binding / Platelet degranulation / protein tyrosine kinase activity / protease binding / calmodulin binding / non-specific serine/threonine protein kinase / phosphorylation / protein serine kinase activity / protein serine/threonine kinase activity / calcium ion binding / positive regulation of gene expression / protein kinase binding / enzyme binding / extracellular exosome / extracellular region / ATP binding / identical protein binding / cytosolSimilarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.69 Å | ||||||
 Authors Authors | Mueller, S. / Kursula, I. / Wilmanns, M. | ||||||
 Citation Citation | Journal: To be Published Title: Structure of M1 Domain from Titin Authors: Mueller, S. / Kursula, I. / Wilmanns, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2bk8.cif.gz | 34.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2bk8.ent.gz | 24 KB | Display | PDB format |
| PDBx/mmJSON format | 2bk8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bk/2bk8ftp://data.pdbj.org/pub/pdb/validation_reports/bk/2bk8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 11043.252 Da / Num. of mol.: 1 / Fragment: RESIDUES 25073-25166 (IG-LIKE DOMAIN) Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Tissue: MUSCLESkeletal muscle / Plasmid: PETM11 / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3)References: UniProt: Q8WZ42, Transferases; Transferring phosphorus-containing groups; Phosphotransferases with an alcohol group as acceptor |
|---|---|
| #2: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 129 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 129 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.21 Å3/Da / Density % sol: 44 % |
|---|---|
| Crystal grow | pH: 7 / Details: 2.2 M AMMONIUM SULFATE 0.1 M BIS TRIS PH 7.0 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.8128 / Beamline: X11 / Wavelength: 0.8128 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Nov 16, 2004 / Details: MIRRORS |
| Radiation | Monochromator: TRIANGULAR MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8128 Å / Relative weight: 1 |
| Reflection | Resolution: 1.69→17.74 Å / Num. obs: 24619 / % possible obs: 99.6 % / Observed criterion σ(I): -3 / Redundancy: 3.8 % / Rmerge(I) obs: 0.04 / Net I/σ(I): 18.1 |
| Reflection shell | Resolution: 1.69→1.8 Å / Redundancy: 3.8 % / Rmerge(I) obs: 0.41 / Mean I/σ(I) obs: 3.89 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: HOMOLOGY MODEL Resolution: 1.69→17.74 Å / Cor.coef. Fo:Fc: 0.962 / Cor.coef. Fo:Fc free: 0.939 / SU ML: 0.085 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.108 / ESU R Free: 0.111 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 37.37 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.69→17.74 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|