 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3e78: Structure determination of the cancer-associated Mycoplasma hyorh... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3.0E+78 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure determination of the cancer-associated Mycoplasma hyorhinis protein Mh-p37 | ||||||
 Components Components | High affinity transport system protein p37 | ||||||
 Keywords Keywords | TPP Binding Protein / Mycoplasma / p37 / TPP / Cell membrane / Lipoprotein / Membrane / Palmitate / Transport | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of protein phosphorylation / regulation of cell migration / host cell cytoplasm / plasma membrane Similarity search - Function | ||||||
| Biological species |  Mycoplasma hyorhinis (bacteria) Mycoplasma hyorhinis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SIRAS / Resolution: 1.9 Å X-RAY DIFFRACTION / SIRAS / Resolution: 1.9 Å | ||||||
 Authors Authors | Sippel, K.H. / Robbins, A.H. / Reutzel, R. / Domsic, J. / McKenna, R. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2008 Title: Structure determination of the cancer-associated Mycoplasma hyorhinis protein Mh-p37. Authors: Sippel, K.H. / Robbins, A.H. / Reutzel, R. / Domsic, J. / Boehlein, S.K. / Govindasamy, L. / Agbandje-McKenna, M. / Rosser, C.J. / McKenna, R. #1: Journal: To be PublishedTitle: A magic triangle for experimental phasing of macromolecules Authors: Beck, T. / Krasauskas, A. / Gruene, T. / Sheldrick, G.M. | ||||||
| History |
|
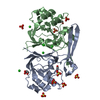
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3e78.cif.gz | 90.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3e78.ent.gz | 68 KB | Display | PDB format |
| PDBx/mmJSON format | 3e78.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/e7/3e78ftp://data.pdbj.org/pub/pdb/validation_reports/e7/3e78 | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 46117.820 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycoplasma hyorhinis (bacteria) / Gene: p37 / Plasmid: pET31p37 / Production host: |
|---|---|
| #2: Chemical | ChemComp-TPP / 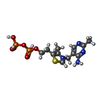  Mass: 425.314 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H19N4O7P2S Mass: 425.314 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H19N4O7P2S |
| #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #4: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 147 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 147 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.15 Å3/Da / Density % sol: 42.69 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: batch / pH: 3 Details: 0.1 M Ammonium Bromide, O.1 M Citric Acid, 40% PEG 4000, 9% N-decyl-beta-D-maltoside detergent , pH 3.0, Batch, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: May 5, 2008 / Details: Osmic mirrors |
| Radiation | Monochromator: Graphite / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→25 Å / Num. obs: 29894 / % possible obs: 96.2 % / Redundancy: 3.5 % / Biso Wilson estimate: 26.427 Å2 / Rsym value: 0.067 / Net I/σ(I): 16.3 |
| Reflection shell | Resolution: 1.9→1.98 Å / Redundancy: 3.5 % / Mean I/σ(I) obs: 5 / Num. unique all: 2860 / Rsym value: 0.376 / % possible all: 93.2 |
- Processing
Processing
| Software |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SIRAS / Resolution: 1.9→25 Å / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / Stereochemistry target values: Engh & Huber / Details: Used weighted full matrix least squares procedure
| |||||||||||||||
| Displacement parameters | Biso mean: 32.694 Å2 | |||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→25 Å
| |||||||||||||||
| Refine LS restraints |
|
