 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3crt | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structural characterization of an engineered allosteric protein | ||||||
 Components Components | Glutathione S-transferase class-mu 26 kDa isozyme | ||||||
 Keywords Keywords | TRANSFERASE / protein design / engineered allostery. pH-switch | ||||||
| Function / homology |  Function and homology information Function and homology informationglutathione transferase / glutathione transferase activity / glutathione metabolic process Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Sagermann, M. / Chapleau, R. / DeLorimier, E. / Lei, M. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2009 Title: Using affinity chromatography to engineer and characterize pH-dependent protein switches. Authors: Sagermann, M. / Chapleau, R.R. / DeLorimier, E. / Lei, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3crt.cif.gz | 60.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3crt.ent.gz | 43.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3crt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cr/3crtftp://data.pdbj.org/pub/pdb/validation_reports/cr/3crt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3cruC  3d0zC  1gneS  3crs S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 25063.148 Da / Num. of mol.: 1 / Mutation: L50C Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|---|
| #2: Chemical | ChemComp-GSH / 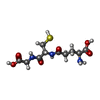  Mass: 307.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N3O6S Mass: 307.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N3O6S |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 158 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 158 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.46 Å3/Da / Density % sol: 49.97 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 20% PEG8000, 50mM tris, 5mM B-mercaptoethanol , pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 150 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: BRUKER AXS MICROSTAR-H / Wavelength: 1.5418 Å |
| Detector | Type: Bruker Platinum 135 / Detector: CCD / Date: Jun 5, 2007 / Details: Helios optics |
| Radiation | Monochromator: Bruker Helios optics / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→19.5 Å / Num. all: 36529 / Num. obs: 34548 / % possible obs: 94.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 9.85 % / Rmerge(I) obs: 0.067 / Net I/σ(I): 20.56 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDBID: 1GNE Resolution: 1.9→19.5 Å / Isotropic thermal model: anisotropic / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: last residues HPPK could not be modeled reliably into the electron density map.
| |||||||||||||||||||||||||
| Displacement parameters |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→19.5 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
|
