 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-334d: DEFINING GC-SPECIFICITY IN THE MINOR GROOVE: SIDE-BY-SIDE BINDING... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 334d | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | DEFINING GC-SPECIFICITY IN THE MINOR GROOVE: SIDE-BY-SIDE BINDING OF THE DI-IMIDAZOLE LEXITROPSIN TO C-A-T-G-G-C-C-A-T-G | ||||||||||||||||||
 Components Components | DNA (5'-D(* Keywords KeywordsDNA / B-DNA / DOUBLE HELIX | Function / homology | DIIMIDAZOLE LEXITROPSIN / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å  Authors AuthorsKopka, M.L. / Goodsell, D.S. / Dickerson, R.E. |  CitationJournal: Structure / Year: 1997 CitationJournal: Structure / Year: 1997Title: Defining GC-specificity in the minor groove: side-by-side binding of the di-imidazole lexitropsin to C-A-T-G-G-C-C-A-T-G. Authors: Kopka, M.L. / Goodsell, D.S. / Han, G.W. / Chiu, T.K. / Lown, J.W. / Dickerson, R.E. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 334d.cif.gz | 26.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb334d.ent.gz | 15.6 KB | Display | PDB format |
| PDBx/mmJSON format | 334d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/34/334dftp://data.pdbj.org/pub/pdb/validation_reports/34/334d | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|





-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 3045.004 Da / Num. of mol.: 2 / Source method: obtained synthetically #2: Chemical | 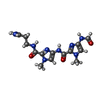  Mass: 362.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H20N9O3 Mass: 362.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H20N9O3#3: Chemical | ChemComp-CA / |   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 62 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 62 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 50 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7.4 Details: pH 7.40, VAPOR DIFFUSION, SITTING DROP, temperature 277.00K | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS Density % sol: 50 % | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 7 | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 278 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Sep 15, 1993 / Details: MIRROR MSC-YALE |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→8 Å / Num. obs: 4895 / % possible obs: 93 % / Biso Wilson estimate: 21.41 Å2 / Rmerge(I) obs: 0.0374 |
| Reflection shell | Resolution: 1.8→1.85 Å / % possible all: 91.9 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 8 Å / % possible obs: 93 % / Observed criterion σ(F): 1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: AN IDEAL HELIX RMS FIT TO BDJ057 Resolution: 1.8→8 Å / σ(F): 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine Biso |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: NUCLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 8 Å / σ(F): 1 / Rfactor obs: 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
