 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2ou2: Acetyltransferase domain of Human HIV-1 Tat interacting protein, ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2ou2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Acetyltransferase domain of Human HIV-1 Tat interacting protein, 60kDa, isoform 3 | ||||||
 Components Components | Histone acetyltransferase HTATIP | ||||||
 Keywords Keywords |  TRANSFERASE / Histone acetyltransferase HTATIP / TIP / TIP60 / Structural Genomics / Structural Genomics Consortium / SGC TRANSFERASE / Histone acetyltransferase HTATIP / TIP / TIP60 / Structural Genomics / Structural Genomics Consortium / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationhistone H2AK5 acetyltransferase activity / peptide crotonyltransferase activity / histone H2A acetyltransferase activity / positive regulation of mitotic sister chromatid segregation / positive regulation of protein acetylation / piccolo histone acetyltransferase complex / Sensing of DNA Double Strand Breaks / MSL complex / peptide butyryltransferase activity / lipid droplet disassembly ...histone H2AK5 acetyltransferase activity / peptide crotonyltransferase activity / histone H2A acetyltransferase activity / positive regulation of mitotic sister chromatid segregation / positive regulation of protein acetylation / piccolo histone acetyltransferase complex / Sensing of DNA Double Strand Breaks / MSL complex / peptide butyryltransferase activity / lipid droplet disassembly / histone H4K16 acetyltransferase activity / sperm DNA condensation / peptide 2-hydroxyisobutyryltransferase activity / positive regulation of attachment of mitotic spindle microtubules to kinetochore / Swr1 complex / positive regulation of triglyceride biosynthetic process / peptidyl-lysine acetylation / histone H4 acetyltransferase activity / regulation of double-strand break repair / Impaired BRCA2 binding to PALB2 / positive regulation of regulatory T cell differentiation / positive regulation of circadian rhythm / positive regulation of innate immune response / DNA repair-dependent chromatin remodeling / Cardiogenesis / Defective homologous recombination repair (HRR) due to BRCA1 loss of function / Defective HDR through Homologous Recombination Repair (HRR) due to PALB2 loss of BRCA1 binding function / Defective HDR through Homologous Recombination Repair (HRR) due to PALB2 loss of BRCA2/RAD51/RAD51C binding function / Homologous DNA Pairing and Strand Exchange / neural tube development / Resolution of D-loop Structures through Synthesis-Dependent Strand Annealing (SDSA) / regulation of hematopoietic stem cell differentiation / Resolution of D-loop Structures through Holliday Junction Intermediates / NuA4 histone acetyltransferase complex / negative regulation of interleukin-2 production / HDR through Single Strand Annealing (SSA) / DNA damage response, signal transduction by p53 class mediator resulting in transcription of p21 class mediator / Impaired BRCA2 binding to RAD51 / peptide-lysine-N-acetyltransferase activity / negative regulation of double-strand break repair via homologous recombination / response to ionizing radiation / positive regulation of double-strand break repair via homologous recombination / mitotic spindle pole / spermatid development / establishment of mitotic spindle orientation / acetyltransferase activity / Presynaptic phase of homologous DNA pairing and strand exchange / positive regulation of myoblast differentiation / cellular response to glucose starvation / positive regulation of autophagy / histone acetyltransferase activity / histone acetyltransferase / Transferases; Acyltransferases; Transferring groups other than aminoacyl groups / neurogenesis / cellular response to estradiol stimulus / nucleotide-excision repair / cellular response to glucose stimulus / transcription coregulator activity / Nonhomologous End-Joining (NHEJ) / double-strand break repair via homologous recombination / Formation of the beta-catenin:TCF transactivating complex / HDR through Homologous Recombination (HRR) / G2/M DNA damage checkpoint / DNA Damage/Telomere Stress Induced Senescence / kinetochore / double-strand break repair / nucleosome / cellular senescence / Recruitment and ATM-mediated phosphorylation of repair and signaling proteins at DNA double strand breaks / site of double-strand break / Processing of DNA double-strand break ends / HATs acetylate histones / proteasome-mediated ubiquitin-dependent protein catabolic process / regulation of apoptotic process / DNA-binding transcription factor binding / Estrogen-dependent gene expression / Regulation of TP53 Activity through Phosphorylation / transcription regulator complex / transcription coactivator activity / regulation of cell cycle / innate immune response / intracellular membrane-bounded organelle / negative regulation of DNA-templated transcription / apoptotic process / DNA damage response / chromatin binding / chromatin / nucleolus / perinuclear region of cytoplasm / positive regulation of DNA-templated transcription / negative regulation of transcription by RNA polymerase II / positive regulation of transcription by RNA polymerase II / nucleoplasm / metal ion binding / nucleus / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Min, J. / Wu, H. / Dombrovski, L. / Loppnau, P. / Weigelt, J. / Sundstrom, M. / Arrowsmith, C.H. / Edwards, A.M. / Bochkarev, A. / Plotnikov, A.N. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: To be Published Title: The Crystal Structure of acetyltransferase domain of Human HIV-1 Tat interacting protein in complex with acetylcoenzyme A. Authors: Wu, H. / Min, J. / Dombrovski, L. / Loppnau, P. / Weigelt, J. / Sundstrom, M. / Arrowsmith, C.H. / Edwards, A.M. / Bochkarev, A. / Plotnikov, A.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2ou2.cif.gz | 68.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2ou2.ent.gz | 48.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2ou2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ou/2ou2ftp://data.pdbj.org/pub/pdb/validation_reports/ou/2ou2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2givS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 32940.055 Da / Num. of mol.: 1 / Fragment: Acetyltransferase domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: HTATIP, TIP60 / Plasmid: Pet28a_MHL / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: Q92993, histone acetyltransferase Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: Q92993, histone acetyltransferase |
|---|---|
| #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn |
| #3: Chemical | ChemComp-ACO / Acetyl-CoA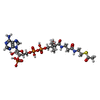  Mass: 809.571 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H38N7O17P3S Mass: 809.571 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H38N7O17P3S |
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 30 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 30 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.94 Å3/Da / Density % sol: 58.14 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.6 Details: Purified HTATIP was complexed with acetylcoenzyme A (AcCoA) (Sigma) at 1:10 molar ratio of protein:AcCoA and crystallized using the hanging drop vapor diffusion method at 20 deg C by mixing ...Details: Purified HTATIP was complexed with acetylcoenzyme A (AcCoA) (Sigma) at 1:10 molar ratio of protein:AcCoA and crystallized using the hanging drop vapor diffusion method at 20 deg C by mixing 1 microliter of the protein solution with 1 microliter of the reservoir solution containing 16% PEG3350, 0.2 M ammonium acetate, 0.1 M Bis-Tris, pH 6.6., VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 1 Å / Beamline: 17-ID / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Sep 20, 2006 |
| Radiation | Monochromator: si111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→83.92 Å / Num. all: 17598 / Num. obs: 17598 / % possible obs: 98.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 5.5 % / Rmerge(I) obs: 0.059 / Rsym value: 0.059 / Net I/σ(I): 9.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 2GIV Resolution: 2.3→83.92 Å / Cor.coef. Fo:Fc: 0.936 / Cor.coef. Fo:Fc free: 0.924 / SU B: 6.075 / SU ML: 0.151 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.263 / ESU R Free: 0.212 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 36.852 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→83.92 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.36 Å / Total num. of bins used: 20
|
