 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1rms: CRYSTAL STRUCTURES OF RIBONUCLEASE MS COMPLEXED WITH 3'-GUANYLIC ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1rms | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURES OF RIBONUCLEASE MS COMPLEXED WITH 3'-GUANYLIC ACID A GP*C ANALOGUE, 2'-DEOXY-2'-FLUOROGUANYLYL-3',5'-CYTIDINE | ||||||
 Components Components | RIBONUCLEASE MS | ||||||
 Keywords Keywords | HYDROLASE(ENDORIBONUCLEASE) | ||||||
| Function / homology |  Function and homology information Function and homology information ribonuclease T1 activity / ribonuclease T1 / endonuclease activity / lyase activity / RNA binding ribonuclease T1 activity / ribonuclease T1 / endonuclease activity / lyase activity / RNA bindingSimilarity search - Function | ||||||
| Biological species |  | ||||||
| Method | X-RAY DIFFRACTION / Resolution: 1.9 Å | ||||||
 Authors Authors | Nonaka, T. / Mitsui, Y. / Nakamura, K.T. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1993 Title: Crystal structure of ribonuclease Ms (as a ribonuclease T1 homologue) complexed with a guanylyl-3',5'-cytidine analogue. Authors: Nonaka, T. / Nakamura, K.T. / Uesugi, S. / Ikehara, M. / Irie, M. / Mitsui, Y. #1: Journal: FEBS Lett. / Year: 1991Title: Three-Dimensional Structure of Ribonuclease Ms(Asterisk)3'-Guanylic Acid Complex at 2.5A Resolution Authors: Nonaka, T. / Mitsui, Y. / Irie, M. / Nakamura, K.T. #2: Journal: J.Mol.Biol. / Year: 1989Title: Crystallization of a Complex between Ribonuclease Ms and 3'-Guanylic Acid Authors: Mitsui, T.Nonaka Y. / Nakamura, K.T. / Watanabe, H. / Ohgi, K. / Irie, M. #3: Journal: J.Biol.Chem. / Year: 1988Title: Three-Dimensional Structure of the Ribonuclease T1(Asterisk)2'-Gmp Complex at 1.9-A Resolution Authors: Arni, R. / Heinemann, U. / Tokuoka, R. / Saenger, W. | ||||||
| History |
| ||||||
| Remark 700 | SHEET ASP 83 (POSITION 1), GLU 84 (POSITION 2) AND ASN 80 (POSITION X) FORM A "G1 TYPE" BETA BULGE. ...SHEET ASP 83 (POSITION 1), GLU 84 (POSITION 2) AND ASN 80 (POSITION X) FORM A "G1 TYPE" BETA BULGE. GLU 84 AND ASN 80 ALSO OCCUPY RESPECTIVELY POSITIONS 0 AND X-1 OF A "WIDE TYPE" BETA BULGE (SEE REMARK 9). ALA 86 (POSITION 1), GLY 87 (POSITION 2) AND ILE 78 (POSITION X) FORM A "CLASSIC TYPE" BETA BULGE. ALA 86 AND ILE 78 ALSO OCCUPY POSITIONS 3 AND X+1 OF A "WIDE TYPE" BETA BULGE (SEE REMARK 9). GLU 4 (POSITION 1), TYR 5 (POSITION 2) AND TYR 12 (POSITION X) FORM A "CLASSIC TYPE" BETA BULGE. GLU 84 (POSITION 0), LEU 85 (POSITION 1) ALA 86 (POSITION 3) ILE 78 (POSITION X-1), PHE 79 (POSITION X) AND ASN 80 (POSITION X+1) FORM A "WIDE TYPE" BETA BULGE (SEE REMARKS 6 AND 7). |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1rms.cif.gz | 29.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1rms.ent.gz | 22.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1rms.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rm/1rmsftp://data.pdbj.org/pub/pdb/validation_reports/rm/1rms | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUE 38 IS A CIS PROLINE. 2: THE PEPTIDE LINKAGE GLY 53-THR 54 HAS BEEN FOUND TO BE IN THE CIS-CONFORMATION AFTER LEAST-SQUARES REFINEMENT. |
-Components
| #1: Protein | Mass: 11409.980 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-3GP / Guanosine monophosphate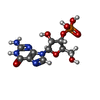  Mass: 363.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O8P Mass: 363.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O8P |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 24 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 24 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.45 Å3/Da / Density % sol: 49.9 % |
|---|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 1.9 Å / Num. obs: 9223 / Num. measured all: 10035 / Rmerge(I) obs: 0.116 |
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.9→6 Å Details: THE RMS DEVIATION BETWEEN THE SUPERIMPOSED CA POSITIONS OF RNASE MS AND RNASE T1 (REFERENCE 3, PROTEIN DATA BANK ENTRY 1RNT) EXCLUDING THE FOUR N-TERMINAL RESIDUES IS 0.55 ANGSTROMS. ...Details: THE RMS DEVIATION BETWEEN THE SUPERIMPOSED CA POSITIONS OF RNASE MS AND RNASE T1 (REFERENCE 3, PROTEIN DATA BANK ENTRY 1RNT) EXCLUDING THE FOUR N-TERMINAL RESIDUES IS 0.55 ANGSTROMS. SIGNIFICANT DEVIATION OCCURS ONLY AT THE N-TERMINUS EXCEPT FOR A LOOP-OUT PORTION WHICH INDICATES THAT THE T1 SEQUENCE, GLY 34-SER 35-ASN 36-SER 37, SHOULD BE ALIGNED WITH THE MS SEQUENCE ASP-35-GAP-GAP-ASP 36. THE FOLLOWING CRITERIA WERE ADOPTED FOR ASSIGNING MAIN-CHAIN HYDROGEN BONDS: THE MAXIMUM DISTANCE BETWEEN AN AMIDE HYDROGEN AND A CARBONYL OXYGEN IS 2.60 ANGSTROMS. THE MAXIMUM DISTANCE BETWEEN AN AMIDE NITROGEN AND A CARBONYL OXYGEN IS 3.35.ANGSTROMS. THE MINIMUM NHO ANGLE IS 100 DEGREES. THE MINIMUM COH ANGLE IS 100 DEGREES. THE MINIMUM CON ANGLE IS 100 DEGREES. HYDROGEN ATOM POSITIONS WERE CALCULATED USING THE PROGRAM *X-PLOR*. THE ASSIGNMENT OF BETA-TURN TYPES IS BASED ON WILMOT AND THORNTON (C.M.WILMOT AND J.M.THORNTON, PROTEIN ENGINEERING, 3, 479-493, 1990). PHI,PSI ANGLES ARE ALLOWED TO DEVIATE BY 30 DEGREES FROM THEIR IDEAL VALUES.
| ||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→6 Å
| ||||||||||||
| Refine LS restraints |
|
