 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1itu | ||||||
|---|---|---|---|---|---|---|---|
| Title | HUMAN RENAL DIPEPTIDASE COMPLEXED WITH CILASTATIN | ||||||
 Components Components | RENAL DIPEPTIDASE | ||||||
 Keywords Keywords | HYDROLASE / DIPEPTIDASE / GLYCOPROTEIN / MEMBRANE-BOUND / ZINC PROTEASE BETA-LACTAMASE / CILASTATIN / COMPLEX (HYDROLASE-INHIBITOR) | ||||||
| Function / homology |  Function and homology information Function and homology informationlactam catabolic process / leukotriene D4 catabolic process / membrane dipeptidase / antibiotic metabolic process / GPI anchor binding / LTC4-CYSLTR mediated IL4 production / glutathione catabolic process / modified amino acid binding / homocysteine metabolic process / Aflatoxin activation and detoxification ...lactam catabolic process / leukotriene D4 catabolic process / membrane dipeptidase / antibiotic metabolic process / GPI anchor binding / LTC4-CYSLTR mediated IL4 production / glutathione catabolic process / modified amino acid binding / homocysteine metabolic process / Aflatoxin activation and detoxification / Synthesis of Leukotrienes (LT) and Eoxins (EX) / metallodipeptidase activity / dipeptidase activity / metalloexopeptidase activity / microvillus membrane / side of membrane / neutrophil chemotaxis / glutathione metabolic process / cellular response to nitric oxide / cellular response to calcium ion / negative regulation of cell migration / beta-lactamase activity / beta-lactamase / apical part of cell / cell junction / apical plasma membrane / inflammatory response / proteolysis / extracellular space / extracellular exosome / zinc ion binding / nucleoplasm / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Nitanai, Y. / Satow, Y. / Adachi, H. / Tsujimoto, M. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2002 Title: Crystal Structure of Human Renal Dipeptidase Involved in beta-Lactam Hydrolysis Authors: Nitanai, Y. / Satow, Y. / Adachi, H. / Tsujimoto, M. #1: Journal: J.CRYST.GROWTH / Year: 1996Title: Crystallization and preliminary X-ray investigation of a glycoprotein, human renal dipeptidase Authors: Nitanai, Y. / Satow, Y. / Adachi, H. / Tsujimoto, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1itu.cif.gz | 170.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1itu.ent.gz | 132.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1itu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/it/1ituftp://data.pdbj.org/pub/pdb/validation_reports/it/1itu | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a dimer generated from the dimer in the asymmetric unit. |
-Components
| #1: Protein | Mass: 41108.195 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Tissue: renal cortex / Plasmid: PHILD2 / Production host:  Pichia pastoris (fungus) / Strain (production host): GS115 / References: UniProt: P16444, membrane dipeptidase Pichia pastoris (fungus) / Strain (production host): GS115 / References: UniProt: P16444, membrane dipeptidase#2: Sugar | ChemComp-NAG /   Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 4 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #3: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn#4: Chemical | 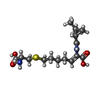  Mass: 358.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H26N2O5S Mass: 358.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H26N2O5S#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 607 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 607 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 3 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.21 Å3/Da / Density % sol: 44.5 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 283 K / Method: vapor diffusion, hanging drop / pH: 7.6 Details: PEG 8000, HEPES, pH 7.6, VAPOR DIFFUSION, HANGING DROP, temperature 283.0K | ||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 10 ℃ / pH: 5.6 / Details: Nitanai, Y., (1996) J.CRYST.GROWTH, 168, 280. | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.5418 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Nov 30, 1996 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→20 Å / Num. all: 46290 / Num. obs: 46290 / % possible obs: 96.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 4 % / Biso Wilson estimate: 16.19 Å2 / Rmerge(I) obs: 0.087 / Net I/σ(I): 13.4 |
| Reflection shell | Resolution: 2→2.05 Å / Rmerge(I) obs: 0.274 / % possible all: 84.5 |
| Reflection | *PLUS Lowest resolution: 20 Å / Num. measured all: 185396 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: NATIVE DIPEPTIDASE STRUCTURE Resolution: 2→10 Å / Data cutoff high absF: 1000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.22 Å2 | ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→10 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.05 Å / Total num. of bins used: 15
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
