 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1fro | ||||||
|---|---|---|---|---|---|---|---|
| Title | HUMAN GLYOXALASE I WITH BENZYL-GLUTATHIONE INHIBITOR | ||||||
 Components Components | LACTOYLGLUTATHIONE LYASE | ||||||
 Keywords Keywords | LACTOYLGLUTATHIONE LYASE / GLYOXALASE I | ||||||
| Function / homology |  Function and homology information Function and homology informationlactoylglutathione lyase / lactoylglutathione lyase activity / methylglyoxal metabolic process / Pyruvate metabolism / glutathione metabolic process / osteoclast differentiation / carbohydrate metabolic process / regulation of transcription by RNA polymerase II / negative regulation of apoptotic process / extracellular exosome ...lactoylglutathione lyase / lactoylglutathione lyase activity / methylglyoxal metabolic process / Pyruvate metabolism / glutathione metabolic process / osteoclast differentiation / carbohydrate metabolic process / regulation of transcription by RNA polymerase II / negative regulation of apoptotic process / extracellular exosome / zinc ion binding / nucleoplasm / plasma membrane / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MIR / Resolution: 2.2 Å X-RAY DIFFRACTION / MIR / Resolution: 2.2 Å | ||||||
 Authors Authors | Cameron, A.D. / Jones, T.A. | ||||||
 Citation Citation | Journal: EMBO J. / Year: 1997 Title: Crystal structure of human glyoxalase I--evidence for gene duplication and 3D domain swapping. Authors: Cameron, A.D. / Olin, B. / Ridderstrom, M. / Mannervik, B. / Jones, T.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1fro.cif.gz | 158.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1fro.ent.gz | 127.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1fro.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fr/1froftp://data.pdbj.org/pub/pdb/validation_reports/fr/1fro | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||
| 2 | 
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 20672.520 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Plasmid: PKK223-3 / Production host:  #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-GSB / 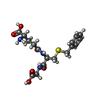  Mass: 397.446 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C17H23N3O6S Mass: 397.446 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C17H23N3O6S#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 360 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 360 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.37 Å3/Da / Density % sol: 42 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.8 / Details: pH 5.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 15 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Feb 20, 1996 |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→30 Å / Num. obs: 35503 / % possible obs: 91.3 % / Redundancy: 3.3 % / Biso Wilson estimate: 27 Å2 / Rmerge(I) obs: 0.068 / Net I/σ(I): 9 |
| Reflection shell | Resolution: 2.2→2.3 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.248 / Mean I/σ(I) obs: 3 / % possible all: 78.1 |
| Reflection | *PLUS Num. measured all: 118633 |
| Reflection shell | *PLUS % possible obs: 79.1 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR / Resolution: 2.2→7.5 Å / Isotropic thermal model: RESTRAINED INDIVIDUAL Details: THIS STRUCTURE WAS REFINED WITH STRICT NON-CRYSTALLOGRAPHIC SYMMETRY CONSTRAINTS BETWEEN ALL MOLECULES. WATER MOLECULES B 246 AND B 253 ARE INVOLVED IN STEREOCHEMICAL CLASHES ON GENERATION ...Details: THIS STRUCTURE WAS REFINED WITH STRICT NON-CRYSTALLOGRAPHIC SYMMETRY CONSTRAINTS BETWEEN ALL MOLECULES. WATER MOLECULES B 246 AND B 253 ARE INVOLVED IN STEREOCHEMICAL CLASHES ON GENERATION OF THE FULL ASYMMETRIC UNIT. DISORDERED SIDE CHAINS HAVE BEEN INCLUDED WITH OCCUPANCIES OF 0.01. THE DENSITY ASSOCIATED WITH THE LAST THREE RESIDUES IS RATHER POOR AND CONSEQUENTLY THE POSITIONS OF THESE RESIDUES ARE RATHER AMBIGUOUS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.25 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→7.5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: STRICT NCS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.3 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
