 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1b1u | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE BIFUNCTIONAL INHIBITOR RAGI | |||||||||
 Components Components | PROTEIN (ALPHA-AMYLASE/TRYPSIN INHIBITOR RATI) | |||||||||
 Keywords Keywords | HYDROLASE INHIBITOR / ALPHA-AMYLASE/TRYPSIN INHIBITOR (RATI) / BIFUNCTIONAL | |||||||||
| Function / homology |  Function and homology information Function and homology informationalpha-amylase inhibitor activity / serine-type endopeptidase inhibitor activity / extracellular region Similarity search - Function | |||||||||
| Biological species |  Eleusine coracana (finger millet) Eleusine coracana (finger millet) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | |||||||||
 Authors Authors | Gourinath, S. / Srinivasan, A. / Singh, T.P. | |||||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2000 Title: Structure of the bifunctional inhibitor of trypsin and alpha-amylase from ragi seeds at 2.2 A resolution. Authors: Gourinath, S. / Alam, N. / Srinivasan, A. / Betzel, C. / Singh, T.P. #1: Journal: Biochemistry / Year: 1995Title: Determination of the Three-Dimensional Structure of the Bifunctional Alpha-Amylase/Trypsin Inhibitor from Ragi Seeds by NMR Spectroscopy Authors: Strobl, S. / Muhlhahn, P. / Bernstein, R. / Wiltscheck, R. / Maskos, K. / Wunderlich, M. / Huber, R. / Glockshuber, R. / Holak, T.A. #2: Journal: J.Mol.Biol. / Year: 1991Title: Preliminary X-Ray Investigation of a Bifunctional Inhibitor from Indian Finger Millet (Ragi) Authors: Srinivasan, A. / Raman, A. / Singh, T.P. #3: Journal: FEBS Lett. / Year: 1983Title: The Complete Amino Acid Sequence of the Bifunctional Alpha-Amylase/Trypsin Inhibitor from Seeds of Ragi (Indian Finger Millet, Eleusine Coracana Gaertn.) Authors: Campos, F.A.P. / Richardson, M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
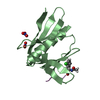
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1b1u.cif.gz | 36.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1b1u.ent.gz | 24.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1b1u.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1b1u_validation.pdf.gz | 362.2 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1b1u_full_validation.pdf.gz | 370.6 KB | Display | |
| Data in XML | 1b1u_validation.xml.gz | 4.8 KB | Display | |
| Data in CIF | 1b1u_validation.cif.gz | 6.8 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b1/1b1uftp://data.pdbj.org/pub/pdb/validation_reports/b1/1b1u | HTTPS FTP |
-Related structure data
| Related structure data |  1bipS S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13153.313 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Eleusine coracana (finger millet) / Organ: SEED / References: UniProt: P01087 |
|---|---|
| #2: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 88 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 88 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.96 Å3/Da / Density % sol: 37.2 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8 Details: 1.15M AMMONIUM SULFATE, 0.2M AMMONIUM PHOSPHATE PH 8.0 | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: microdialysis | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 173 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X31 / Wavelength: 1 / Beamline: X31 / Wavelength: 1 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jun 23, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→20 Å / Num. obs: 5684 / % possible obs: 98.7 % / Redundancy: 12.2 % / Biso Wilson estimate: 25.4 Å2 / Rsym value: 0.094 / Net I/σ(I): 11.2 |
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 4.2 % / Mean I/σ(I) obs: 2.27 / Rsym value: 0.299 / % possible all: 94.6 |
| Reflection | *PLUS Highest resolution: 2.2 Å / Lowest resolution: 10 Å / Num. obs: 5094 / % possible obs: 91 % / Num. measured all: 57852 / Rmerge(I) obs: 0.072 |
| Reflection shell | *PLUS Highest resolution: 2.7 Å / Lowest resolution: 2.79 Å / % possible obs: 83.5 % / Redundancy: 4.2 % / Rmerge(I) obs: 0.162 / Mean I/σ(I) obs: 2.27 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1BIP, MODEL 20 Resolution: 2.2→10 Å / Rfactor Rfree error: 0.023 / Data cutoff high absF: 100000 / Data cutoff low absF: 0.1 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.8 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.3 Å / Luzzati d res low obs: 10 Å / Luzzati sigma a obs: 0.35 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.34 Å / Rfactor Rfree error: 0.083 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Num. reflection obs: 5094 / Rfactor all: 0.224 / Rfactor obs: 0.219 / Rfactor Rfree: 0.277 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.2 Å |
