National Institutes of Health/National Institute Of Allergy and Infectious Diseases (NIH/NIAID)
AI095755
米国
引用
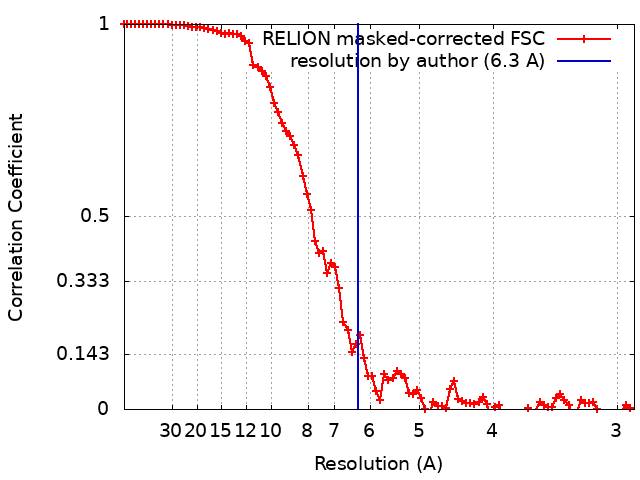
ジャーナル: Nat Microbiol / 年: 2020 タイトル: Structural insights into the transition of Clostridioides difficile binary toxin from prepore to pore. 著者: David M Anderson / Michael J Sheedlo / Jaime L Jensen / D Borden Lacy / 要旨: Clostridioides (formerly Clostridium) difficile is a Gram-positive, spore-forming anaerobe and a leading cause of hospital-acquired infection and gastroenteritis-associated death in US hospitals. The ...Clostridioides (formerly Clostridium) difficile is a Gram-positive, spore-forming anaerobe and a leading cause of hospital-acquired infection and gastroenteritis-associated death in US hospitals. The disease state is usually preceded by disruption of the host microbiome in response to antibiotic treatment and is characterized by mild to severe diarrhoea. C. difficile infection is dependent on the secretion of one or more AB-type toxins: toxin A (TcdA), toxin B (TcdB) and the C. difficile transferase toxin (CDT). Whereas TcdA and TcdB are considered the primary virulence factors, recent studies suggest that CDT increases the severity of C. difficile infection in some of the most problematic clinical strains. To better understand how CDT functions, we used cryo-electron microscopy to define the structure of CDTb, the cell-binding component of CDT. We obtained structures of several oligomeric forms that highlight the conformational changes that enable conversion from a prepore to a β-barrel pore. The structural analysis also reveals a glycan-binding domain and residues involved in binding the host-cell receptor, lipolysis-stimulated lipoprotein receptor. Together, these results provide a framework to understand how CDT functions at the host cell interface.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 マップデータ
マップデータ 試料
試料 キーワード
キーワード Clostridium (クロストリジウム属) /
Clostridium (クロストリジウム属) /  機能・相同性情報
機能・相同性情報
 データ登録者
データ登録者 米国, 2件
米国, 2件  引用
引用 構造の表示
構造の表示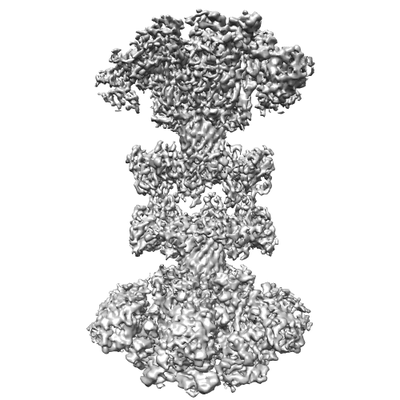
 ダウンロードとリンク
ダウンロードとリンク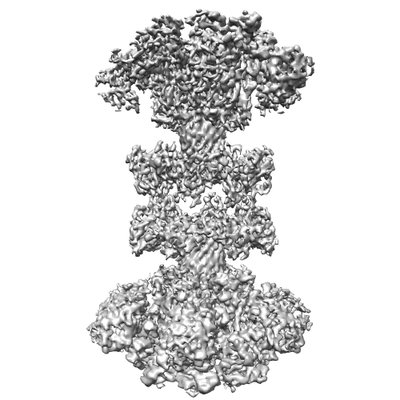 emd_0608.png
emd_0608.png http://ftp.pdbj.org/pub/emdb/structures/EMD-0608
http://ftp.pdbj.org/pub/emdb/structures/EMD-0608

















 試料の構成要素
試料の構成要素 解析
解析 電子顕微鏡法
電子顕微鏡法