登録情報 データベース : EMDB / ID : EMD-2595タイトル Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome Reconstruction of Intermediate state (s2). 試料 : 26S Proteasome from Saccharomyces cerevisiaeタンパク質・ペプチド : 26S Proteasome / / / 機能・相同性 分子機能 ドメイン・相同性 構成要素
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / 生物種 Saccharomyces cerevisiae (パン酵母)手法 / / 解像度 : 9.3 Å Unverdorben P / Beck F / Sledz P / Schweitzer A / Pfeifer G / Plitzko JM / Baumeister W / Foerster F ジャーナル : Proc Natl Acad Sci U S A / 年 : 2014タイトル : Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome.著者 : Pia Unverdorben / Florian Beck / Paweł Śledź / Andreas Schweitzer / Günter Pfeifer / Jürgen M Plitzko / Wolfgang Baumeister / Friedrich Förster / 要旨 : The 26S proteasome is a 2.5 MDa molecular machine that executes the degradation of substrates of the ubiquitin-proteasome pathway. The molecular architecture of the 26S proteasome was recently ... The 26S proteasome is a 2.5 MDa molecular machine that executes the degradation of substrates of the ubiquitin-proteasome pathway. The molecular architecture of the 26S proteasome was recently established by cryo-EM approaches. For a detailed understanding of the sequence of events from the initial binding of polyubiquitylated substrates to the translocation into the proteolytic core complex, it is necessary to move beyond static structures and characterize the conformational landscape of the 26S proteasome. To this end we have subjected a large cryo-EM dataset acquired in the presence of ATP and ATP-γS to a deep classification procedure, which deconvolutes coexisting conformational states. Highly variable regions, such as the density assigned to the largest subunit, Rpn1, are now well resolved and rendered interpretable. Our analysis reveals the existence of three major conformations: in addition to the previously described ATP-hydrolyzing (ATPh) and ATP-γS conformations, an intermediate state has been found. Its AAA-ATPase module adopts essentially the same topology that is observed in the ATPh conformation, whereas the lid is more similar to the ATP-γS bound state. Based on the conformational ensemble of the 26S proteasome in solution, we propose a mechanistic model for substrate recognition, commitment, deubiquitylation, and translocation into the core particle. 履歴 登録 2014年2月25日 - ヘッダ(付随情報) 公開 2014年3月26日 - マップ公開 2014年4月2日 - 更新 2016年2月17日 - 現状 2016年2月17日 処理サイト : PDBe / 状態 : 公開
すべて表示 表示を減らす
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 マップデータ
マップデータ 試料
試料 キーワード
キーワード Proteasome (プロテアソーム) /
Proteasome (プロテアソーム) /  機能・相同性情報
機能・相同性情報
 データ登録者
データ登録者 引用
引用
 構造の表示
構造の表示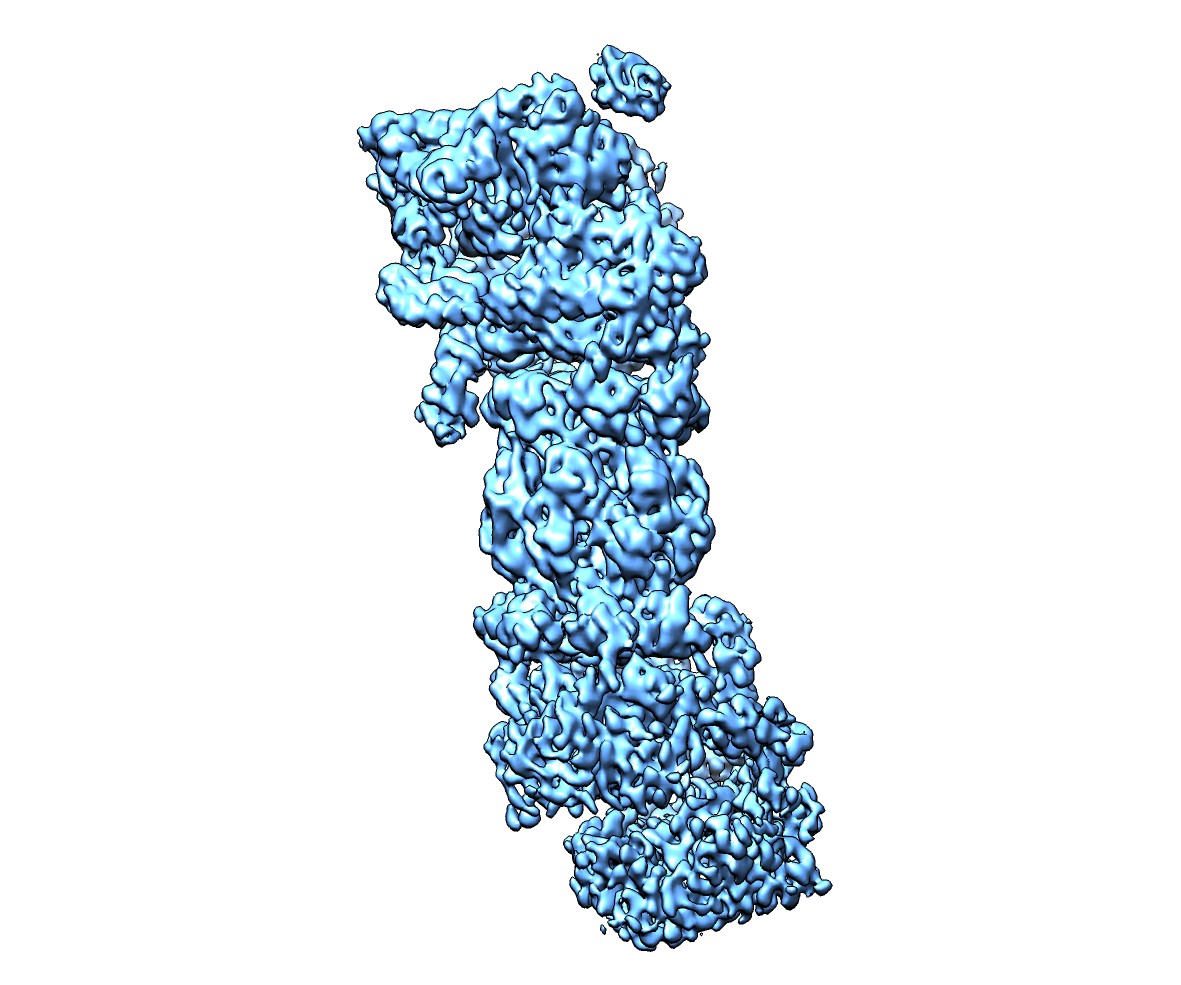
 ダウンロードとリンク
ダウンロードとリンク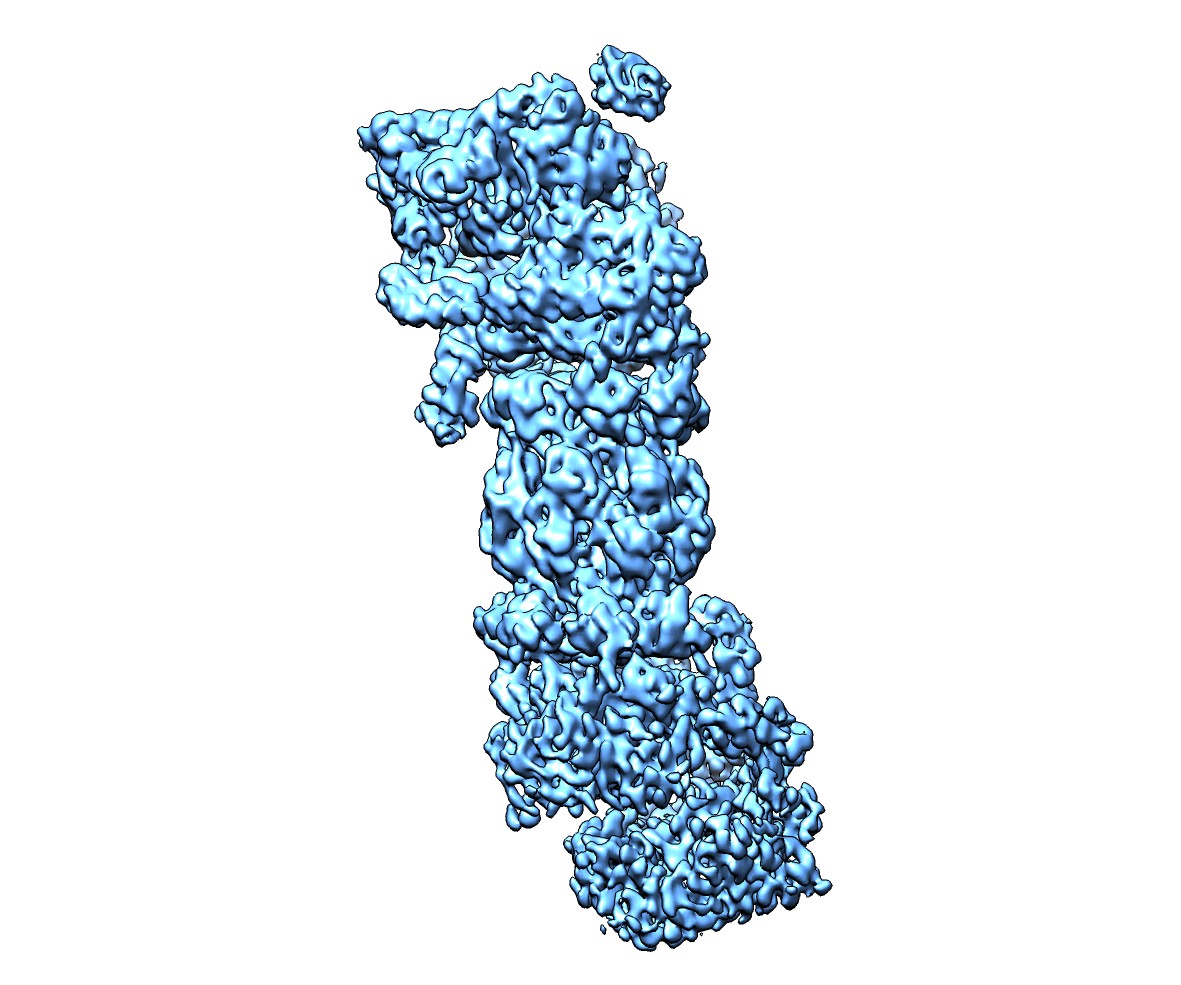 emd_2595.jpg
emd_2595.jpg http://ftp.pdbj.org/pub/emdb/structures/EMD-2595
http://ftp.pdbj.org/pub/emdb/structures/EMD-2595
































 試料の構成要素
試料の構成要素 解析
解析 電子顕微鏡法
電子顕微鏡法
