 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9t4m: Human Diphosphoinositol Polyphosphate Phosphohydrolase 1 (DIPP1) ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9t4m | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Human Diphosphoinositol Polyphosphate Phosphohydrolase 1 (DIPP1) N112S mutant in complex with IP6 | |||||||||
 Components Components | Diphosphoinositol polyphosphate phosphohydrolase 1 | |||||||||
 Keywords Keywords | HYDROLASE / Inositol metabolism / phosphatase / NUDIX hydrolases / DIPP1 / IP6 | |||||||||
| Function / homology |  Function and homology information Function and homology informationinositol diphosphate pentakisphosphate diphosphatase activity / diphosphoinositol polyphosphate catabolic process / inositol diphosphate tetrakisphosphate diphosphatase activity / endopolyphosphatase / diadenosine polyphosphate catabolic process / 5'-(N7-methyl 5'-triphosphoguanosine)-[mRNA] diphosphatase / bis(5'-adenosyl)-hexaphosphatase activity / inositol-3,5-bisdiphosphate-2,3,4,6-tetrakisphosphate 5-diphosphatase activity / diadenosine pentaphosphate catabolic process / diadenosine hexaphosphate catabolic process ...inositol diphosphate pentakisphosphate diphosphatase activity / diphosphoinositol polyphosphate catabolic process / inositol diphosphate tetrakisphosphate diphosphatase activity / endopolyphosphatase / diadenosine polyphosphate catabolic process / 5'-(N7-methyl 5'-triphosphoguanosine)-[mRNA] diphosphatase / bis(5'-adenosyl)-hexaphosphatase activity / inositol-3,5-bisdiphosphate-2,3,4,6-tetrakisphosphate 5-diphosphatase activity / diadenosine pentaphosphate catabolic process / diadenosine hexaphosphate catabolic process / adenosine 5'-(hexahydrogen pentaphosphate) catabolic process / endopolyphosphatase activity / diphosphoinositol polyphosphate metabolic process / diphosphoinositol-polyphosphate diphosphatase activity / inositol-5-diphosphate-1,2,3,4,6-pentakisphosphate diphosphatase activity / 5'-(N(7)-methyl 5'-triphosphoguanosine)-[mRNA] diphosphatase activity / diadenosine hexaphosphate hydrolase (ATP-forming) / bis(5'-adenosyl)-pentaphosphatase activity / Synthesis of pyrophosphates in the cytosol / diphosphoinositol-polyphosphate diphosphatase / RNA decapping / 5'-(N7-methylguanosine 5'-triphospho)-[mRNA] hydrolase / 5'-(N(7)-methylguanosine 5'-triphospho)-[mRNA] hydrolase activity / manganese ion binding / cell-cell signaling / glutamatergic synapse / magnesium ion binding / zinc ion binding / nucleus / cytoplasm / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.78 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.78 Å | |||||||||
 Authors Authors | Casas-Florez, D. / Marquez-Monino, M.A. / Gonzalez, B. | |||||||||
| Funding support |  Spain, 2items Spain, 2items
| |||||||||
 Citation Citation | Journal: Int.J.Biol.Macromol. / Year: 2026 Title: The DIPP1 family binds IP 8 in catalytically-productive twist-boat and chair conformations and associates in a ligand-dependent manner. Authors: Casas-Florez, D. / Whitfield, H. / Perez-Canadillas, J.M. / Monterroso, B. / Riley, A.M. / Marquez-Monino, M.A. / Shipton, M.L. / Sanz-Aparicio, J. / Brearley, C.A. / Potter, B.V.L. / Gonzalez, B. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9t4m.cif.gz | 57.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9t4m.ent.gz | 38.6 KB | Display | PDB format |
| PDBx/mmJSON format | 9t4m.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/t4/9t4mftp://data.pdbj.org/pub/pdb/validation_reports/t4/9t4m | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  9t48C  9t49C  9t4aC  9t4bC  9t4cC  9t4dC  9t4eC  9t4fC  9t4gC  9t4hC  9t4iC  9t4jC  9t4kC  9t4lC C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 17088.342 Da / Num. of mol.: 1 Mutation: Deletion of the last 24 C-terminal residues (149-172) and N112S Source method: isolated from a genetically manipulated source Details: Deletion of the last 24 C-terminal residues (149-172), the first three amino acids (GHM) remain as residual residues after removal of the expression tag. Source: (gene. exp.) Homo sapiens (human) / Gene: NUDT3, DIPP, DIPP1 / Production host:  References: UniProt: O95989, diphosphoinositol-polyphosphate diphosphatase, diadenosine hexaphosphate hydrolase (ATP-forming), endopolyphosphatase, 5'-(N7-methylguanosine 5'-triphospho)-[mRNA] ...References: UniProt: O95989, diphosphoinositol-polyphosphate diphosphatase, diadenosine hexaphosphate hydrolase (ATP-forming), endopolyphosphatase, 5'-(N7-methylguanosine 5'-triphospho)-[mRNA] hydrolase, 5'-(N7-methyl 5'-triphosphoguanosine)-[mRNA] diphosphatase |
|---|
-Non-polymers , 5 types, 200 molecules 
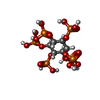



| #2: Chemical |  Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2#3: Chemical | ChemComp-IHP / |  Mass: 660.035 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H18O24P6 / Feature type: SUBJECT OF INVESTIGATION Mass: 660.035 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H18O24P6 / Feature type: SUBJECT OF INVESTIGATION#4: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#5: Chemical |  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 193 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.9 Å3/Da / Density % sol: 58.5 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, sitting drop / pH: 5 Details: Protein: 24mg/mL Ip6: 10mM Precipitant condition: 31% PEG 6K, 0.1M NaOAc pH 5, 0.2M LiCl, 10mM MgCl2. Ratio: 1:1 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: MASSIF-3 / Wavelength: 0.9677 Å / Beamline: MASSIF-3 / Wavelength: 0.9677 Å |
| Detector | Type: DECTRIS EIGER X 4M / Detector: PIXEL / Date: Jun 13, 2023 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9677 Å / Relative weight: 1 |
| Reflection | Resolution: 1.78→50.07 Å / Num. obs: 19071 / % possible obs: 100 % / Redundancy: 15.9 % / Biso Wilson estimate: 18.6 Å2 / CC1/2: 0.99 / Rpim(I) all: 0.13 / Net I/σ(I): 4.5 |
| Reflection shell | Resolution: 1.78→1.81 Å / Mean I/σ(I) obs: 0.1 / Num. unique obs: 943 / CC1/2: 0.33 / Rpim(I) all: 6.95 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 1.78→50.07 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.918 / SU B: 3.427 / SU ML: 0.103 / Cross valid method: FREE R-VALUE / ESU R: 0.122 / ESU R Free: 0.128 Details: Hydrogens have been added in their riding positions
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.887 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.78→50.07 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
