 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9qh3: Pseudomonas aeruginosa polynucleotide phosphorylase in complex wi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9qh3 | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Pseudomonas aeruginosa polynucleotide phosphorylase in complex with recognition site of RNase E | ||||||||||||||||||||||||
 Components Components |
| ||||||||||||||||||||||||
 Keywords Keywords | RNA BINDING PROTEIN / polynucleotide phosphorylase / ribonuclease E / RNA degradosome | ||||||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationribonuclease E / ribonuclease E activity / polyribonucleotide nucleotidyltransferase / polyribonucleotide nucleotidyltransferase activity / RNA catabolic process / tRNA processing / mRNA catabolic process / RNA nuclease activity / RNA processing / cytoplasmic side of plasma membrane ...ribonuclease E / ribonuclease E activity / polyribonucleotide nucleotidyltransferase / polyribonucleotide nucleotidyltransferase activity / RNA catabolic process / tRNA processing / mRNA catabolic process / RNA nuclease activity / RNA processing / cytoplasmic side of plasma membrane / rRNA processing / 3'-5'-RNA exonuclease activity / tRNA binding / rRNA binding / magnesium ion binding / RNA binding / zinc ion binding / cytosol / cytoplasm Similarity search - Function | ||||||||||||||||||||||||
| Biological species |  Pseudomonas aeruginosa PAO1 (bacteria) Pseudomonas aeruginosa PAO1 (bacteria) | ||||||||||||||||||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 2.4 Å | ||||||||||||||||||||||||
 Authors Authors | Paris, G. / Luisi, B.F. | ||||||||||||||||||||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||||||||||||||||||||
 Citation Citation | Journal: Nucleic Acids Res / Year: 2025 Title: A multi-dentate, cooperative interaction between endo- and exo-ribonucleases within the bacterial RNA degradosome. Authors: Giulia Paris / Kai Katsuya-Gaviria / Hannah Clarke / Margaret Johncock / Tom Dendooven / Aleksei Lulla / Ben F Luisi / Abstract: In Escherichia coli and numerous other bacteria, two of the principal enzymes mediating messenger RNA decay and RNA processing-RNase E, an endoribonuclease, and polynucleotide phosphorylase (PNPase), ...In Escherichia coli and numerous other bacteria, two of the principal enzymes mediating messenger RNA decay and RNA processing-RNase E, an endoribonuclease, and polynucleotide phosphorylase (PNPase), an exoribonuclease-assemble into a multi-enzyme complex known as the RNA degradosome. While RNase E forms a homotetramer and PNPase a homotrimer, it remains unclear how these two enzymes interact within the RNA degradosome to potentially satisfy all mutual recognition sites. In this study, we used cryo-EM, biochemistry, and biophysical studies to discover and characterize a new binding mode for PNPase encompassing two or more motifs that are necessary and sufficient for strong interaction with RNase E. While a similar interaction is seen in Salmonella enterica, a different recognition mode arose for Pseudomonas aeruginosa, illustrating the evolutionary drive to maintain physical association of the two ribonucleases. The data presented here suggest a model for the quaternary organization of the RNA degradosome of E. coli, where one PNPase trimer interacts with one RNase E protomer. Conformational transitions are predicted to facilitate substrate capture and transfer to catalytic centres. The model suggests how the endo- and exo-ribonucleases might cooperate in cellular RNA turnover and recruitment of regulatory RNA by the degradosome assembly. | ||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9qh3.cif.gz | 599.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9qh3.ent.gz | 500.2 KB | Display | PDB format |
| PDBx/mmJSON format | 9qh3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qh/9qh3ftp://data.pdbj.org/pub/pdb/validation_reports/qh/9qh3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  53153MC  9qh0C M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |

-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 59727.867 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Details: catalytic core, without S1 and KH domains / Source: (gene. exp.) Pseudomonas aeruginosa PAO1 (bacteria) / Gene: pnp, PA4740 / Production host: References: UniProt: Q9HV59, polyribonucleotide nucleotidyltransferase #2: Protein/peptide | | Mass: 2911.264 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: PNPase recognition site from RNase E / Source: (gene. exp.) Pseudomonas aeruginosa PAO1 (bacteria) / Gene: rne, PA2976 / Production host: #3: Chemical | 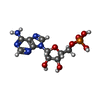  Type: RNA linking / Mass: 347.221 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H14N5O7P / Feature type: SUBJECT OF INVESTIGATION Type: RNA linking / Mass: 347.221 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H14N5O7P / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 45 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 45 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: Polynucleotide phosphorylase in complex with recognition site from ribonuclease E Type: COMPLEX Details: Complex prepared by co-expression and chromatographic purification Entity ID: #1-#2 / Source: RECOMBINANT |
|---|---|
| Molecular weight | Experimental value: NO |
| Source (natural) | Organism:  Pseudomonas aeruginosa (bacteria) / Strain: PAO1 Pseudomonas aeruginosa (bacteria) / Strain: PAO1 |
| Source (recombinant) | Organism: |
| Buffer solution | pH: 8 Details: 20 mM Tris-HCl pH 8.0, 25 mM MgCl2, 150 mM KCl, 1 mM TCEP |
| Specimen | Conc.: 3.6 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES Details: Purified PNPase core and RNE-muGFP-CHis proteins were mixed in 1:2 ratio and their complex were separated on the Superdex 200 Increase 10/300 GL column (Cytiva) equilibrated with Cryo-EM ...Details: Purified PNPase core and RNE-muGFP-CHis proteins were mixed in 1:2 ratio and their complex were separated on the Superdex 200 Increase 10/300 GL column (Cytiva) equilibrated with Cryo-EM buffer (20 mM Tris-HCl pH 8.0, 25 mM MgCl2, 150 mM KCl, 1 mM TCEP). Peak fractions were combined, the protein was concentrated to 15 microM using Amicon Ultra concentrator with 10 kDa cut-off (Millipore) and used to prepare Cryo-EM grids. The samples were mixed with CHAPSO (3-([3-cholamidopropyl]dimethylammonio)-2-hydroxy-1-propanesulfonate) at a final concentration of 8 mM |
| Vitrification | Instrument: FEI VITROBOT MARK IV / Cryogen name: ETHANE / Humidity: 95 % / Chamber temperature: 277.2 K / Details: blotting force -4, 3 sec blot time |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: TFS KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Nominal defocus max: 1800 nm / Nominal defocus min: 600 nm |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Image recording | Average exposure time: 4.39 sec. / Electron dose: 53.94 e/Å2 / Film or detector model: FEI FALCON IV (4k x 4k) / Num. of grids imaged: 1 / Num. of real images: 4000 |
| EM imaging optics | Energyfilter name: TFS Selectris |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: NONE | ||||||||||||||||||||||||
| Symmetry | Point symmetry: C1 (asymmetric) | ||||||||||||||||||||||||
| 3D reconstruction | Resolution: 2.4 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 95237 / Symmetry type: POINT | ||||||||||||||||||||||||
| Atomic model building | Protocol: AB INITIO MODEL / Space: REAL / Details: phenix refine and manual rebuilding using COOT | ||||||||||||||||||||||||
| Atomic model building | Source name: AlphaFold / Type: in silico model | ||||||||||||||||||||||||
| Refinement | Highest resolution: 2.4 Å Stereochemistry target values: REAL-SPACE (WEIGHTED MAP SUM AT ATOM CENTERS) | ||||||||||||||||||||||||
| Refine LS restraints |
|
