 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9q6w: Crystal Structure of Vibrio cholerae PilU, a PilT-dependent Retra... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9q6w | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Vibrio cholerae PilU, a PilT-dependent Retraction ATPase - Crystal Form 2 | ||||||
 Components Components | Twitching motility protein PilT | ||||||
 Keywords Keywords | MOTOR PROTEIN / Structural Genomics / Center for Structural Biology of Infectious Diseases / CSBID / PilU / PilT-dependent Retraction ATPase | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA endonuclease activity, producing 5'-phosphomonoesters / type IV pilus-dependent motility / ATP hydrolysis activity / ATP binding Similarity search - Function | ||||||
| Biological species |  Vibrio cholerae O1 biovar El Tor str. N16961 (bacteria) Vibrio cholerae O1 biovar El Tor str. N16961 (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å | ||||||
 Authors Authors | Minasov, G. / Shukla, S. / Shuvalova, L. / Brunzelle, J.S. / Satchell, K.J.F. / Center for Structural Biology of Infectious Diseases (CSBID) | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Protein Sci / Year: 2026 Title: Analysis of the heterogenous structural states of the hexameric ATPase PilU of the type IV pili from Vibrio cholerae. Authors: Yirui Guo / Shantanu Shukla / George Minasov / Nicole L Inniss / Thomas Klose / Valerie L Tokars / Alfonso Mondragón / Zbyszek Otwinowski / Dominika Borek / Karla J F Satchell / Abstract: Type IV pili (T4P) mediate surface motility, host interactions, and DNA uptake through cycles of extension and retraction. While the primary retraction ATPase PilT has been extensively characterized, ...Type IV pili (T4P) mediate surface motility, host interactions, and DNA uptake through cycles of extension and retraction. While the primary retraction ATPase PilT has been extensively characterized, its homolog PilU remains less well understood despite being demonstrated as a PilT-dependent retraction ATPase. Here, we determined six PilU structures by cryo-electron microscopy and x-ray crystallography. The structures reveal a homohexameric assembly stabilized by interactions between the C-terminal catalytic domain of one subunit and the N-terminal PAS-like domain of a neighboring subunit. PilU adopts multiple conformational states, exhibiting different combinations of open and closed interfaces even in the absence of nucleotide. Comparison with PilT highlights structural features that likely underlie PilU's weak ATPase activity and its dependence on PilT for function. Together, these findings provide a structural framework for understanding PilU's role within the T4P retraction machinery. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9q6w.cif.gz | 425.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9q6w.ent.gz | 352.1 KB | Display | PDB format |
| PDBx/mmJSON format | 9q6w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q6/9q6wftp://data.pdbj.org/pub/pdb/validation_reports/q6/9q6w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  10myC  10mzC  10naC  10nbC  9q6mC C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |




-Links
 PDBj
PDBj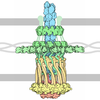
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 42949.852 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Vibrio cholerae O1 biovar El Tor str. N16961 (bacteria)Gene: VC_0463 / Plasmid: pET-15b / Production host: #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 55 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 55 / Source method: obtained synthetically / Formula: SO4#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 128 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 128 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | Has protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.1 Å3/Da / Density % sol: 41.6 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: Protein: 15.0 mg/ml, 0.5M Sodium chloride, 1mM TCEP; Screen: AmSO4 (B1), 0.2 M Cadmium sulfate, 2.2 M Ammonium sulfate; Cryo: 2M Lithium sulfate. |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 21-ID-F / Wavelength: 0.97872 Å |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Mar 15, 2023 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97872 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→30 Å / Num. obs: 30441 / % possible obs: 100 % / Observed criterion σ(I): -3 / Redundancy: 7.5 % / Biso Wilson estimate: 41.6 Å2 / CC1/2: 0.996 / CC star: 0.999 / Rmerge(I) obs: 0.141 / Rpim(I) all: 0.056 / Rrim(I) all: 0.152 / Rsym value: 0.141 / Χ2: 0.949 / Net I/σ(I): 16.1 |
| Reflection shell | Resolution: 2.7→2.75 Å / Redundancy: 7.5 % / Mean I/σ(I) obs: 1.8 / Num. unique obs: 1485 / CC1/2: 0.697 / CC star: 0.906 / Rpim(I) all: 0.537 / Χ2: 0.92 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.7→29.58 Å / Cor.coef. Fo:Fc: 0.917 / Cor.coef. Fo:Fc free: 0.87 / SU B: 32.623 / SU ML: 0.329 / Cross valid method: THROUGHOUT / ESU R Free: 0.435 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 48.83 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.7→29.58 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|