 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9o02: Crystal structure of AfOgg1-K122S mutant bound to 8-OG DNA duplex... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9o02 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of AfOgg1-K122S mutant bound to 8-OG DNA duplex in an intermediate state | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | HYDROLASE / LYASE/DNA / DNA-glycosylase / AP-lyase / LYASE-DNA complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationhydrolase activity, hydrolyzing N-glycosyl compounds / DNA-(apurinic or apyrimidinic site) lyase / class I DNA-(apurinic or apyrimidinic site) endonuclease activity / base-excision repair / Hydrolases; Glycosylases; Hydrolysing N-glycosyl compounds Similarity search - Function | |||||||||
| Biological species |   Archaeoglobus fulgidus (archaea) Archaeoglobus fulgidus (archaea)synthetic construct (others) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | |||||||||
 Authors Authors | Huffman, J.L. / Syed, A. / Tang, H.Y.H. / Arvai, A.S. / Mol, C.D. / Tainer, J.A. | |||||||||
| Funding support |  United States, 2items United States, 2items
| |||||||||
 Citation Citation | Journal: Nat Commun / Year: 2026 Title: A unified catalytic mechanism in bifunctional DNA glycosylases with an evolutionarily conserved aspartate-lysine dyad. Authors: Syed, A. / Serafim, L.F. / Arvai, A.S. / Minko, I.G. / Tang, H.Y.H. / Huffman, J.L. / Mol, C.D. / Hitomi, K. / Sarker, A.H. / Parikh, S. / Tsai, C.L. / Bacolla, A. / Shin, D.S. / Cunningham, ...Authors: Syed, A. / Serafim, L.F. / Arvai, A.S. / Minko, I.G. / Tang, H.Y.H. / Huffman, J.L. / Mol, C.D. / Hitomi, K. / Sarker, A.H. / Parikh, S. / Tsai, C.L. / Bacolla, A. / Shin, D.S. / Cunningham, R.P. / Iwai, S. / Chowdhury, D. / Lloyd, R.S. / Ivanov, I. / Tainer, J.A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9o02.cif.gz | 71.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9o02.ent.gz | 46.2 KB | Display | PDB format |
| PDBx/mmJSON format | 9o02.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/o0/9o02ftp://data.pdbj.org/pub/pdb/validation_reports/o0/9o02 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  10mnC  9nz8C  9nz9C  9nzaC  9nzcC  9nzdC  9o00C  9o01C  9o03C C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23125.824 Da / Num. of mol.: 1 / Mutation: K122S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Archaeoglobus fulgidus (archaea) / Gene: ogg, AF_0371 / Production host:  References: UniProt: O29876, Hydrolases; Glycosylases; Hydrolysing N-glycosyl compounds, DNA-(apurinic or apyrimidinic site) lyase |
|---|---|
| #2: DNA chain | Mass: 4609.999 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) |
| #3: DNA chain | Mass: 4584.984 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) |
| #4: Chemical | ChemComp-MOO / 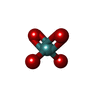  Mass: 159.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: MoO4 Mass: 159.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: MoO4 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.15 Å3/Da / Density % sol: 42.79 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop Details: 20-30% PEG4000, 100 mM MES, pH 6.5, 5% potassium permanganate (saturated) |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL / Beamline: BL11-1 / Wavelength: 0.9648 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Oct 7, 2002 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9648 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→52.05 Å / Num. obs: 9608 / % possible obs: 99.9 % / Redundancy: 4.1 % / CC1/2: 0.998 / Rmerge(I) obs: 0.083 / Net I/σ(I): 7.6 |
| Reflection shell | Resolution: 2.5→2.54 Å / Num. unique obs: 468 / CC1/2: 0.436 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.5→52.05 Å / SU ML: 0.35 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 33.99 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→52.05 Å
| |||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
