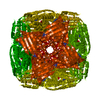
Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9m2r: Imidazole glycerol phosphate dehydratase from Mycobacterium tuber... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9m2r | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Imidazole glycerol phosphate dehydratase from Mycobacterium tuberculosis, apo structure | |||||||||||||||
 Components Components | Imidazoleglycerol-phosphate dehydratase | |||||||||||||||
 Keywords Keywords | LYASE / histidine pathway / dehydratase / Mycobacterium tuberculosis | |||||||||||||||
| Function / homology |  Function and homology information Function and homology informationimidazoleglycerol-phosphate dehydratase / imidazoleglycerol-phosphate dehydratase activity / L-histidine biosynthetic process / metal ion binding / cytoplasm Similarity search - Function | |||||||||||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria) | |||||||||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 2.2 Å | |||||||||||||||
 Authors Authors | Raina, R. / Kar, D. / Singla, M. / Tiwari, S. / Kumari, S. / Aneja, S. / Kumar, V. / Banerjee, S. / Goyal, S. / Pal, R.K. ...Raina, R. / Kar, D. / Singla, M. / Tiwari, S. / Kumari, S. / Aneja, S. / Kumar, V. / Banerjee, S. / Goyal, S. / Pal, R.K. / Vinothkumar, K.R. / Biswal, B.K. | |||||||||||||||
| Funding support |  India, 4items India, 4items
| |||||||||||||||
 Citation Citation | Journal: Acta Crystallogr F Struct Biol Commun / Year: 2025 Title: Cryo-EM structures of Mycobacterium tuberculosis imidazole glycerol phosphate dehydratase in the apo state and in the presence of small molecules. Authors: Rahul Raina / Deepsikha Kar / Mohini Singla / Satish Tiwari / Swati Kumari / Sonanjali Aneja / Varun Kumar / Soumya Banerjee / Shivika Goyal / Ravi Kant Pal / Kutti R Vinothkumar / Bichitra Biswal / Abstract: Unlike humans, Mycobacterium tuberculosis (Mtb), the causative agent of human tuberculosis, has a de novo histidine-biosynthesis pathway. The enzyme imidazole glycerol phosphate dehydratase (IGPD), ...Unlike humans, Mycobacterium tuberculosis (Mtb), the causative agent of human tuberculosis, has a de novo histidine-biosynthesis pathway. The enzyme imidazole glycerol phosphate dehydratase (IGPD), which catalyses the conversion of imidazole glycerol phosphate to imidazole acetol phosphate, has been studied extensively from various organisms and has become a major target for the development of antibacterial, antiweed and antifungal small molecules. In our previous studies, we have shown that in crystals IGPD forms a 24-mer oligomeric state in which the monomers are arranged in 432 symmetry. In order to gain insights into the oligomeric state of Mtb IGPD in solution, we determined cryogenic sample electron microscopy (cryo-EM) structures of apo IGPD at 2.2 and 3.1 Å resolution. In addition, we also determined the cryo-EM structure of IGPD in the presence of 3-amino-1,2,4-triazole (ATZ) to 2.8 Å resolution. The results of this work, which corroborate those from the crystallographic studies, indicate that IGPD forms a homo-oligomeric structure in solution comprising of 24 subunits. ATZ binds in the active-site pocket of the enzyme, which is located at the interface of three monomers and tethers 24 ATZ molecules. The results of this study suggest that cryo-EM, in addition to being a rapidly evolving and complementary imaging technology for elucidating 3D structures of biological macromolecules, can be useful in pinpointing the mode of binding small molecules of low mass (here ∼85 Da) and mapping protein-ligand interactions, which could assist in the design of accurate (high-potency) inhibitors. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9m2r.cif.gz | 879.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9m2r.ent.gz | 723.2 KB | Display | PDB format |
| PDBx/mmJSON format | 9m2r.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/m2/9m2rftp://data.pdbj.org/pub/pdb/validation_reports/m2/9m2r | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  63590MC  9m2pC  9m2qC C: citing same article ( M: map data used to model this data |
|---|---|
| Similar structure data |


-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 23633.516 Da / Num. of mol.: 24 Source method: isolated from a genetically manipulated source Details: The enzyme was expressed with a Histag at the N-terminus but not observed in the experiment. The C-terminal residues are also not observed in the experiment. The original uniprot ID doesn't ...Details: The enzyme was expressed with a Histag at the N-terminus but not observed in the experiment. The C-terminal residues are also not observed in the experiment. The original uniprot ID doesn't exist (I6XBW5) but uniparc ID is UPI000012C861. Source: (gene. exp.) Mycobacterium tuberculosis (bacteria) / Gene: hisB, Rv1601, MTCY336.03c / Production host: Mycolicibacterium smegmatis (bacteria) / Strain (production host): MC2 (4517)References: UniProt: P9WML9, imidazoleglycerol-phosphate dehydratase #2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 48 / Source method: obtained synthetically / Formula: Mn / Feature type: SUBJECT OF INVESTIGATION Mass: 54.938 Da / Num. of mol.: 48 / Source method: obtained synthetically / Formula: Mn / Feature type: SUBJECT OF INVESTIGATION#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1032 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1032 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: Mycobacterium tuberculosis HisB,5-bromo-1-propyl-1H-1,2,4-traizole added but not modelled Type: COMPLEX / Details: An oligomer of 24 subunits / Entity ID: #1 / Source: RECOMBINANT | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | Value: 0.55 MDa / Experimental value: NO | |||||||||||||||
| Source (natural) | Organism: Mycobacterium tuberculosis (bacteria) | |||||||||||||||
| Source (recombinant) | Organism: Mycolicibacterium smegmatis (bacteria) / Strain: MC2 (4517) | |||||||||||||||
| Buffer solution | pH: 8.5 | |||||||||||||||
| Buffer component |
| |||||||||||||||
| Specimen | Conc.: 8 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES Details: Before freezing, 0.5% chapso and 100 uM BrATZ (5-bromo-1-propyl-1H-1,2,4-traizole) was added | |||||||||||||||
| Specimen support | Grid material: GOLD / Grid mesh size: 300 divisions/in. / Grid type: Quantifoil R1.2/1.3 | |||||||||||||||
| Vitrification | Instrument: FEI VITROBOT MARK IV / Cryogen name: ETHANE / Humidity: 100 % / Chamber temperature: 291 K |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: TFS KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 96000 X / Calibrated magnification: 166666 X / Nominal defocus max: 35000 nm / Nominal defocus min: 12000 nm / Cs: 2.7 mm / C2 aperture diameter: 50 µm / Alignment procedure: COMA FREE |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Image recording | Average exposure time: 40 sec. / Electron dose: 26.5 e/Å2 / Detector mode: COUNTING / Film or detector model: FEI FALCON III (4k x 4k) / Num. of grids imaged: 1 |
| Image scans | Sampling size: 14 µm / Width: 4096 / Height: 4096 |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Particle selection | Num. of particles selected: 150382 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Point symmetry: O (octahedral) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 2.2 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 96264 / Algorithm: BACK PROJECTION / Num. of class averages: 1 / Symmetry type: POINT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic model building | B value: 103.3 / Protocol: OTHER / Space: RECIPROCAL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic model building | PDB-ID: 4LPF Pdb chain-ID: A / Accession code: 4LPF / Chain residue range: 10-199 / Pdb chain residue range: 10-199 / Source name: PDB / Type: experimental model | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | Resolution: 2.2→141.12 Å / Cor.coef. Fo:Fc: 0.967 / SU B: 3.364 / SU ML: 0.08 / ESU R: 0.173 Stereochemistry target values: MAXIMUM LIKELIHOOD WITH PHASES Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: PARAMETERS FOR MASK CACLULATION | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 101.769 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Total: 36336 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|