 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9krf: Alpha-hemolysin heptameric pore state bound to 10:0 PC lipid chai... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9krf | ||||||
|---|---|---|---|---|---|---|---|
| Title | Alpha-hemolysin heptameric pore state bound to 10:0 PC lipid chains derived from 10:0 PC liposomes | ||||||
 Components Components | Alpha-hemolysin | ||||||
 Keywords Keywords | LIPID BINDING PROTEIN / PFT / Liposomes | ||||||
| Function / homology |  Function and homology information Function and homology informationcytolysis in another organism / The NLRP3 inflammasome / Purinergic signaling in leishmaniasis infection / toxin activity / extracellular region / identical protein binding Similarity search - Function | ||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 2.8 Å | ||||||
 Authors Authors | Chatterjee, A. / Roy, A. / Dutta, S. | ||||||
| Funding support |  India, 1items India, 1items
| ||||||
 Citation Citation | Journal: Nat Commun / Year: 2025 Title: Structural insights into pre-pore intermediates of alpha-hemolysin in the lipidic environment. Authors: Arnab Chatterjee / Anupam Roy / Thejas Satheesh / Partho Pratim Das / Bapan Mondal / Prithiv Kishore / Mahipal Ganji / Somnath Dutta / Abstract: The infectious microbe Staphylococcus aureus releases an array of cytotoxic pore-forming toxins (PFTs) that severely damage the cell membrane during bacterial infection. However, the interaction ...The infectious microbe Staphylococcus aureus releases an array of cytotoxic pore-forming toxins (PFTs) that severely damage the cell membrane during bacterial infection. However, the interaction interfaces between the host cell membrane and toxin were hardly explored. So far, there are no pore, and intermediate structures of these toxins available in the presence of bio-membrane, which could elucidate the pore-forming mechanism. Here, we investigate the structure of different conformational states of this alpha-hemolysin (α-HL/Hla), a β-PFT in lipidic environment using single-particle cryo-EM. Additionally, we explore lipid destabilization by the toxin, using single-molecule imaging, confocal imaging, and validation of lipid-protein interactions using mutational studies. We elucidate eight cryo-EM structures of wildtype α-HL with various liposomes, which are composed of either 10:0 PC or Egg-PC/Cholesterol or Egg-PC/Sphingomyelin or 10:0 PC/Sphingomyelin. Our structural and biophysical studies confirm that different chain lengths and various membrane compositions facilitate the formation of intermediate pre-pores and complete pore species. We also demonstrate that the percentage of pre-pore population increases due to sphingomyelin-induced membrane rigidity. Altogether, this study unveils the structure-function analysis of the pre-pore to pore transition of wildtype α-HL during its crosstalk with the lipid membrane. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9krf.cif.gz | 408.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9krf.ent.gz | 338.3 KB | Display | PDB format |
| PDBx/mmJSON format | 9krf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kr/9krfftp://data.pdbj.org/pub/pdb/validation_reports/kr/9krf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  62310MC  9kg0C  9kg1C  9kg3C  9kg6C  9kreC  9ktmC  9ktoC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 33290.000 Da / Num. of mol.: 7 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (bacteria) / Gene: hly, hla / Production host: #2: Chemical | ChemComp-P1O / 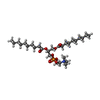  Mass: 566.728 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C28H57NO8P / Feature type: SUBJECT OF INVESTIGATION / Comment: DDPC, phospholipid*YM Mass: 566.728 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C28H57NO8P / Feature type: SUBJECT OF INVESTIGATION / Comment: DDPC, phospholipid*YMHas ligand of interest | Y | Has protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: Alpha-hemolysin heptameric pore state bound to 10:0 PC lipid chains derived from 10:0 PC liposomes Type: COMPLEX / Entity ID: #1 / Source: RECOMBINANT |
|---|---|
| Source (natural) | Organism: Staphylococcus aureus (bacteria) |
| Source (recombinant) | Organism: |
| Buffer solution | pH: 8 |
| Specimen | Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TALOS ARCTICA |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: OTHER FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: OTHER |
| Electron lens | Mode: BRIGHT FIELD / Nominal defocus max: 3500 nm / Nominal defocus min: 750 nm |
| Image recording | Electron dose: 40 e/Å2 / Film or detector model: GATAN K2 SUMMIT (4k x 4k) |
- Processing
Processing
| EM software | Name: PHENIX / Category: model refinement | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||
| 3D reconstruction | Resolution: 2.8 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 127106 / Symmetry type: POINT | ||||||||||||||||||||||||
| Refinement | Highest resolution: 2.8 Å Stereochemistry target values: REAL-SPACE (WEIGHTED MAP SUM AT ATOM CENTERS) | ||||||||||||||||||||||||
| Refine LS restraints |
|
