 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 9hkz | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | Cryo-EM structure of Pseudomonas aeruginosa tetrameric S-adenosyl-L-homocysteine hydrolase with 2 wide open, 1 open and 1 closed subunits | ||||||
 要素 要素 | Adenosylhomocysteinase | ||||||
 キーワード キーワード | HYDROLASE / SAM-DEPENDENT METHYLATION / PROTEIN DYNAMICS / CRYOEM | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報L-homocysteine biosynthetic process / adenosylhomocysteinase / adenosylhomocysteinase activity / L-methionine cycle / one-carbon metabolic process / cytosol 類似検索 - 分子機能 | ||||||
| 生物種 |   Pseudomonas aeruginosa (緑膿菌) Pseudomonas aeruginosa (緑膿菌) | ||||||
| 手法 | 電子顕微鏡法 / 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 3.1 Å | ||||||
 データ登録者 データ登録者 | Malecki, P.H. / Wozniak, K. / Ruszkowski, M. / Brzezinski, K. | ||||||
| 資金援助 |  ポーランド, 1件 ポーランド, 1件
| ||||||
 引用 引用 | ジャーナル: To Be Published タイトル: Cryo-EM structure of Pseudomonas aeruginosa tetrameric S-adenosyl-L-homocysteine hydrolase with 2 wide open, 1 open and 1 closed subunits 著者: Malecki, P.H. / Wozniak, K. / Ruszkowski, M. / Brzezinski, K. #1: ジャーナル: Acta Crystallogr D Struct Biol / 年: 2019 タイトル: Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. 著者: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / ...著者: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / Robert D Oeffner / Billy K Poon / Michael G Prisant / Randy J Read / Jane S Richardson / David C Richardson / Massimo D Sammito / Oleg V Sobolev / Duncan H Stockwell / Thomas C Terwilliger / Alexandre G Urzhumtsev / Lizbeth L Videau / Christopher J Williams / Paul D Adams /    要旨: Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological ...Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological processes and to develop new therapeutics against diseases. The overall structure-solution workflow is similar for these techniques, but nuances exist because the properties of the reduced experimental data are different. Software tools for structure determination should therefore be tailored for each method. Phenix is a comprehensive software package for macromolecular structure determination that handles data from any of these techniques. Tasks performed with Phenix include data-quality assessment, map improvement, model building, the validation/rebuilding/refinement cycle and deposition. Each tool caters to the type of experimental data. The design of Phenix emphasizes the automation of procedures, where possible, to minimize repetitive and time-consuming manual tasks, while default parameters are chosen to encourage best practice. A graphical user interface provides access to many command-line features of Phenix and streamlines the transition between programs, project tracking and re-running of previous tasks. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 9hkz.cif.gz | 382.1 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb9hkz.ent.gz | 284.4 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 9hkz.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/hk/9hkzftp://data.pdbj.org/pub/pdb/validation_reports/hk/9hkz | HTTPS FTP |
|---|
-関連構造データ

-リンク
 PDBj
PDBj
- 集合体
集合体
| 登録構造単位 | 
|
|---|---|
| 1 |
|
-要素
| #1: タンパク質 | 分子量: 51735.031 Da / 分子数: 4 / 由来タイプ: 組換発現 / 由来: (組換発現) Pseudomonas aeruginosa (緑膿菌) / 遺伝子: ahcY, sahH, PA0432 / 発現宿主: #2: 化合物 | ChemComp-NAD /   分子量: 663.425 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C21H27N7O14P2 / コメント: NAD*YM 分子量: 663.425 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C21H27N7O14P2 / コメント: NAD*YM#3: 化合物 | ChemComp-ADN / | 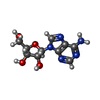  分子量: 267.241 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H13N5O4 分子量: 267.241 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H13N5O4研究の焦点であるリガンドがあるか | N | Has protein modification | N | |
|---|
-実験情報
-実験
| 実験 | 手法: 電子顕微鏡法 |
|---|---|
| EM実験 | 試料の集合状態: PARTICLE / 3次元再構成法: 単粒子再構成法 |
- 試料調製
試料調製
| 構成要素 | 名称: S-adenosyl-L-homocysteine hydrolase / タイプ: COMPLEX / Entity ID: #1 / 由来: RECOMBINANT |
|---|---|
| 分子量 | 値: 0.2 MDa / 実験値: YES |
| 由来(天然) | 生物種: Pseudomonas aeruginosa (緑膿菌) / 株: PAO1 |
| 由来(組換発現) | 生物種: |
| 緩衝液 | pH: 7.5 |
| 試料 | 濃度: 0.5 mg/ml / 包埋: NO / シャドウイング: NO / 染色: NO / 凍結: YES |
| 急速凍結 | 装置: FEI VITROBOT MARK IV / 凍結剤: ETHANE |
- 電子顕微鏡撮影
電子顕微鏡撮影
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
|---|---|
| 顕微鏡 | モデル: TFS KRIOS |
| 電子銃 | 電子線源:  FIELD EMISSION GUN / 加速電圧: 300 kV / 照射モード: FLOOD BEAM FIELD EMISSION GUN / 加速電圧: 300 kV / 照射モード: FLOOD BEAM |
| 電子レンズ | モード: BRIGHT FIELD / 最大 デフォーカス(公称値): 3500 nm / 最小 デフォーカス(公称値): 900 nm / Cs: 2.7 mm |
| 撮影 | 電子線照射量: 40.44 e/Å2 フィルム・検出器のモデル: GATAN K3 BIOQUANTUM (6k x 4k) 撮影したグリッド数: 1 / 実像数: 6384 |
- 解析
解析
| EMソフトウェア | 名称: PHENIX / バージョン: 1.21.2_5419+SVN / カテゴリ: モデル精密化 |
|---|---|
| CTF補正 | タイプ: NONE |
| 対称性 | 点対称性: C1 (非対称) |
| 3次元再構成 | 解像度: 3.1 Å / 解像度の算出法: FSC 0.143 CUT-OFF / 粒子像の数: 172337 / クラス平均像の数: 1 / 対称性のタイプ: POINT |
| 原子モデル構築 | プロトコル: FLEXIBLE FIT / 空間: REAL |
| 原子モデル構築 | PDB-ID: 8CFB Accession code: 8CFB / Source name: PDB / タイプ: experimental model |
| 精密化 | 交差検証法: NONE |

