 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9g82: CTX-M-14 mixed with piperacillin at 3s delay time - serial crysta... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9g82 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | CTX-M-14 mixed with piperacillin at 3s delay time - serial crystallography temperature series; 37C, 310K | |||||||||||||||
 Components Components | Beta-lactamase | |||||||||||||||
 Keywords Keywords | HYDROLASE / catalytic activity / beta-lactamase activity / hydrolase activity | |||||||||||||||
| Function / homology |  Function and homology information Function and homology informationbeta-lactam antibiotic catabolic process / beta-lactamase activity / beta-lactamase / response to antibiotic Similarity search - Function | |||||||||||||||
| Biological species |  Klebsiella pneumoniae (bacteria) Klebsiella pneumoniae (bacteria) | |||||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | |||||||||||||||
 Authors Authors | Prester, A. / von Stetten, D. / Mehrabi, P. / Schulz, E.C. | |||||||||||||||
| Funding support |  Germany, European Union, 4items Germany, European Union, 4items
| |||||||||||||||
 Citation Citation | Journal: Nat Commun / Year: 2025 Title: Probing the modulation of enzyme kinetics by multi-temperature, time-resolved serial crystallography. Authors: Schulz, E.C. / Prester, A. / von Stetten, D. / Gore, G. / Hatton, C.E. / Bartels, K. / Leimkohl, J.P. / Schikora, H. / Ginn, H.M. / Tellkamp, F. / Mehrabi, P. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9g82.cif.gz | 158.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9g82.ent.gz | 103.4 KB | Display | PDB format |
| PDBx/mmJSON format | 9g82.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 9g82_validation.pdf.gz | 1.1 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 9g82_full_validation.pdf.gz | 1.1 MB | Display | |
| Data in XML | 9g82_validation.xml.gz | 17 KB | Display | |
| Data in CIF | 9g82_validation.cif.gz | 23.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g8/9g82ftp://data.pdbj.org/pub/pdb/validation_reports/g8/9g82 | HTTPS FTP |
-Related structure data
| Related structure data |  9g5sC  9g5wC  9g5xC  9g61C  9g6lC  9g6mC  9g6nC  9g6oC  9g6pC  9g7vC  9g7wC  9g7xC  9g7yC  9g7zC  9g80C  9g81C  9i7lC C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 31009.291 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: Residue 1- 27 signal peptide / Source: (gene. exp.) Klebsiella pneumoniae (bacteria) / Gene: blaCTX-M-14, bla_2, SAMEA3512100_05103 / Production host: |
|---|---|
| #2: Chemical | ChemComp-WPP /   Mass: 517.555 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H27N5O7S / Feature type: SUBJECT OF INVESTIGATION Mass: 517.555 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H27N5O7S / Feature type: SUBJECT OF INVESTIGATION |
| #3: Chemical | ChemComp-YPP / 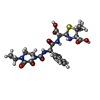  Mass: 535.570 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H29N5O8S / Feature type: SUBJECT OF INVESTIGATION Mass: 535.570 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H29N5O8S / Feature type: SUBJECT OF INVESTIGATION |
| #4: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 174 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 174 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.16 Å3/Da / Density % sol: 43.12 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: batch mode / pH: 4.5 Details: CTX-M-14 solution (22 mg/ml) was mixed with 45% precipitant solution (40% PEG8000, 200mM lithium sulfate, 100mM sodium acetate, pH 4.5) and with 5% undiluted seed stock in batch crystallization setups |
-Data collection
| Diffraction | Mean temperature: 310 K / Ambient temp details: environmental control box / Serial crystal experiment: Y |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, EMBL c/o DESY / Beamline: P14 (MX2) / Wavelength: 0.976 Å |
| Detector | Type: DECTRIS EIGER X 4M / Detector: PIXEL / Date: Sep 15, 2022 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.976 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→78.07 Å / Num. obs: 27982 / % possible obs: 100 % / Redundancy: 174.5 % / Biso Wilson estimate: 24.78 Å2 / CC1/2: 0.959 / CC star: 0.99 / Net I/σ(I): 4.36 |
| Reflection shell | Resolution: 1.7→1.76 Å / Redundancy: 106.4 % / Mean I/σ(I) obs: 0.66 / Num. unique obs: 2734 / CC1/2: 0.252 / CC star: 0.634 / % possible all: 100 |
| Serial crystallography sample delivery | Method: fixed target |
| Serial crystallography sample delivery fixed target | Sample holding: Hare-Chip , silicon |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.7→78.07 Å / SU ML: 0.2651 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 23.5523 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 33.14 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→78.07 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -4.72341321924 Å / Origin y: 1.32292654122 Å / Origin z: 19.8489645416 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
