Journal: FEBS J / Year: 2025 Title: Bacterial transcriptional repressor NrdR - a flexible multifactorial nucleotide sensor. Authors: Inna Rozman Grinberg / Ornella Bimaï / Saher Shahid / Lena Philipp / Markel Martínez-Carranza / Ipsita Banerjee / Daniel Lundin / Pål Stenmark / Britt-Marie Sjöberg / Derek T Logan / Abstract: NrdR is a bacterial transcriptional repressor consisting of a zinc (Zn)-ribbon domain followed by an ATP-cone domain. Understanding its mechanism of action could aid the design of novel ...NrdR is a bacterial transcriptional repressor consisting of a zinc (Zn)-ribbon domain followed by an ATP-cone domain. Understanding its mechanism of action could aid the design of novel antibacterials. NrdR binds specifically to two "NrdR boxes" upstream of ribonucleotide reductase operons, of which Escherichia coli has three: nrdHIEF, nrdDG and nrdAB, in the last of which we identified a new box. We show that E. coli NrdR (EcoNrdR) has similar binding strength to all three sites when loaded with ATP plus deoxyadenosine triphosphate (dATP) or equivalent diphosphate combinations. No other combination of adenine nucleotides promotes binding to DNA. We present crystal structures of EcoNrdR-ATP-dATP and EcoNrdR-ADP-dATP, which are the first high-resolution crystal structures of an NrdR. We have also determined cryo-electron microscopy structures of DNA-bound EcoNrdR-ATP-dATP and novel filaments of EcoNrdR-ATP. Tetrameric forms of EcoNrdR involve alternating interactions between pairs of Zn-ribbon domains and ATP-cones. The structures reveal considerable flexibility in relative orientation of ATP-cones vs Zn-ribbon domains. The structure of DNA-bound EcoNrdR-ATP-dATP shows that significant conformational rearrangements between ATP-cones and Zn-ribbons accompany DNA binding while the ATP-cones retain the same relative orientation. In contrast, ATP-loaded EcoNrdR filaments show rearrangements of the ATP-cone pairs and sequester the DNA-binding residues of NrdR such that they are unable to bind to DNA. Our results, in combination with a previous structural and biochemical study, point to highly flexible EcoNrdR structures that, when loaded with the correct nucleotides, adapt to an optimal promoter-binding conformation.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Sweden,
Sweden,  United States, 6items
United States, 6items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



 PDBj
PDBj


 Assembly
Assembly


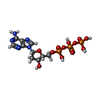




 Mass: 491.182 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O12P3 / Feature type: SUBJECT OF INVESTIGATION
Mass: 491.182 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O12P3 / Feature type: SUBJECT OF INVESTIGATION Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Feature type: SUBJECT OF INVESTIGATION / Comment: ADP, energy-carrying molecule*YM
Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Feature type: SUBJECT OF INVESTIGATION / Comment: ADP, energy-carrying molecule*YM Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION Sample preparation
Sample preparation / Beamline: I04 / Wavelength: 0.9795 Å
/ Beamline: I04 / Wavelength: 0.9795 Å Processing
Processing