 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9e3b | ||||||
|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of PRMT5/WDR77 in complex with 6S complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE / PRMT5 / Methyl transferase / WDR77 / arginine | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of adenylate cyclase-inhibiting dopamine receptor signaling pathway / peptidyl-arginine N-methylation / type II protein arginine methyltransferase / protein-arginine omega-N symmetric methyltransferase activity / peptidyl-arginine methylation / negative regulation of epithelial cell proliferation involved in prostate gland development / Golgi ribbon formation / oocyte axis specification / secretory columnal luminar epithelial cell differentiation involved in prostate glandular acinus development / histone H4R3 methyltransferase activity ...positive regulation of adenylate cyclase-inhibiting dopamine receptor signaling pathway / peptidyl-arginine N-methylation / type II protein arginine methyltransferase / protein-arginine omega-N symmetric methyltransferase activity / peptidyl-arginine methylation / negative regulation of epithelial cell proliferation involved in prostate gland development / Golgi ribbon formation / oocyte axis specification / secretory columnal luminar epithelial cell differentiation involved in prostate glandular acinus development / histone H4R3 methyltransferase activity / epithelial cell proliferation involved in prostate gland development / 7-methylguanosine cap hypermethylation / U12-type spliceosomal complex / protein-arginine N-methyltransferase activity / U1 snRNP binding / pICln-Sm protein complex / methylosome / positive regulation of mRNA splicing, via spliceosome / small nuclear ribonucleoprotein complex / SMN-Sm protein complex / spliceosomal tri-snRNP complex / commitment complex / U2-type precatalytic spliceosome / methyl-CpG binding / mRNA cis splicing, via spliceosome / U2-type prespliceosome assembly / U2-type catalytic step 2 spliceosome / cell volume homeostasis / U2-type spliceosomal complex / endothelial cell activation / U1 snRNP / U2 snRNP / U4 snRNP / chloride transport / histone H3 methyltransferase activity / precatalytic spliceosome / histone methyltransferase activity / regulation of mitotic nuclear division / negative regulation of gene expression via chromosomal CpG island methylation / mRNA Splicing - Minor Pathway / E-box binding / Cul4B-RING E3 ubiquitin ligase complex / histone methyltransferase complex / positive regulation of oligodendrocyte differentiation / spliceosomal complex assembly / U5 snRNP / negative regulation of cell differentiation / regulation of ERK1 and ERK2 cascade / ribonucleoprotein complex binding / spliceosomal snRNP assembly / U4/U6 x U5 tri-snRNP complex / ubiquitin-like ligase-substrate adaptor activity / catalytic step 2 spliceosome / mRNA Splicing - Major Pathway / liver regeneration / RNA splicing / regulation of signal transduction by p53 class mediator / methyltransferase activity / spliceosomal complex / circadian regulation of gene expression / mRNA splicing, via spliceosome / DNA-templated transcription termination / Regulation of TP53 Activity through Methylation / RMTs methylate histone arginines / protein polyubiquitination / p53 binding / transcription corepressor activity / microtubule cytoskeleton / snRNP Assembly / ubiquitin-dependent protein catabolic process / SARS-CoV-2 modulates host translation machinery / transcription coactivator activity / chromatin remodeling / protein heterodimerization activity / positive regulation of cell population proliferation / regulation of transcription by RNA polymerase II / regulation of DNA-templated transcription / chromatin / Golgi apparatus / RNA binding / nucleoplasm / identical protein binding / nucleus / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 3.06 Å | ||||||
 Authors Authors | Jin, C.Y. / Hunkeler, M. / Fischer, E.S. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: J Biol Chem / Year: 2025 Title: Substrate adaptors are flexible tethering modules that enhance substrate methylation by the arginine methyltransferase PRMT5. Authors: Cyrus Y Jin / Moritz Hunkeler / Kathleen M Mulvaney / William R Sellers / Eric S Fischer / Abstract: Protein arginine methyltransferase (PRMT) 5 is an essential arginine methyltransferase responsible for the majority of cellular symmetric dimethyl-arginine marks. PRMT5 uses substrate adaptors such ...Protein arginine methyltransferase (PRMT) 5 is an essential arginine methyltransferase responsible for the majority of cellular symmetric dimethyl-arginine marks. PRMT5 uses substrate adaptors such as pICln, RIOK1, and COPR5 to recruit and methylate a wide range of substrates. Although the substrate adaptors play important roles in substrate recognition, how they direct PRMT5 activity towards specific substrates remains incompletely understood. Using biochemistry and cryogenic electron microscopy, we show that these adaptors compete for the same binding site on PRMT5. We find that substrate adaptor and substrate complexes are bound to PRMT5 through two peptide motifs, enabling these adaptors to act as flexible tethering modules to enhance substrate methylation. Taken together, our results shed structural and mechanistic light on the PRMT5 substrate adaptor function and the biochemical nature of PRMT5 interactors. #1: Journal: Acta Crystallogr D Struct Biol / Year: 2019 Title: Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Authors: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty ...Authors: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / Robert D Oeffner / Billy K Poon / Michael G Prisant / Randy J Read / Jane S Richardson / David C Richardson / Massimo D Sammito / Oleg V Sobolev / Duncan H Stockwell / Thomas C Terwilliger / Alexandre G Urzhumtsev / Lizbeth L Videau / Christopher J Williams / Paul D Adams /   Abstract: Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological ...Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological processes and to develop new therapeutics against diseases. The overall structure-solution workflow is similar for these techniques, but nuances exist because the properties of the reduced experimental data are different. Software tools for structure determination should therefore be tailored for each method. Phenix is a comprehensive software package for macromolecular structure determination that handles data from any of these techniques. Tasks performed with Phenix include data-quality assessment, map improvement, model building, the validation/rebuilding/refinement cycle and deposition. Each tool caters to the type of experimental data. The design of Phenix emphasizes the automation of procedures, where possible, to minimize repetitive and time-consuming manual tasks, while default parameters are chosen to encourage best practice. A graphical user interface provides access to many command-line features of Phenix and streamlines the transition between programs, project tracking and re-running of previous tasks. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9e3b.cif.gz | 1.7 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9e3b.ent.gz | 1.1 MB | Display | PDB format |
| PDBx/mmJSON format | 9e3b.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/e3/9e3bftp://data.pdbj.org/pub/pdb/validation_reports/e3/9e3b | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  47477MC  9e3aC  9e3cC C: citing same article ( M: map data used to model this data |
|---|---|
| Similar structure data |


-Links
 PDBj
PDBj


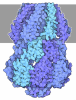


- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 72766.664 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PRMT5, HRMT1L5, IBP72, JBP1, SKB1 / Production host:  Trichoplusia ni (cabbage looper) / Strain (production host): Hi5 Trichoplusia ni (cabbage looper) / Strain (production host): Hi5References: UniProt: O14744, type II protein arginine methyltransferase #2: Protein | Mass: 37186.695 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: WDR77, MEP50, WD45, HKMT1069, Nbla10071 / Production host: Trichoplusia ni (cabbage looper) / References: UniProt: Q9BQA1#3: Protein | Mass: 26552.504 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CLNS1A, CLCI, ICLN / Production host:  #4: Protein | Mass: 13954.288 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: SNRPD1 / Production host: #5: Chemical | ChemComp-SFG / 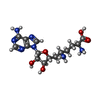  Mass: 381.387 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C15H23N7O5 / Feature type: SUBJECT OF INVESTIGATION Mass: 381.387 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C15H23N7O5 / Feature type: SUBJECT OF INVESTIGATIONHas ligand of interest | Y | Has protein modification | N | Sequence details | The precise residue numbers of the amino acid residues in chains M, N, O, and P (snRNP Sm D1) ...The precise residue numbers of the amino acid residues in chains M, N, O, and P (snRNP Sm D1) cannot be determined because of the number of GR repeats near the C-terminus of the protein. They are numbered such that they end with ARG-114. However, this region could actually be any stretch of GRGRGR between residues 97 and 114 (or in the case of chain N, any stretch of RGRGR between residues 98 and 114). | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component |
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | Value: 0.77 MDa / Experimental value: NO | ||||||||||||||||||||||||||||||||||||||||||
| Source (natural) |
| ||||||||||||||||||||||||||||||||||||||||||
| Source (recombinant) |
| ||||||||||||||||||||||||||||||||||||||||||
| Buffer solution | pH: 7.4 Details: 30 mM HEPES pH 7.4, 150 mM NaCl, 3 mM TCEP, 3.33 mM sinefungin | ||||||||||||||||||||||||||||||||||||||||||
| Buffer component |
| ||||||||||||||||||||||||||||||||||||||||||
| Specimen | Conc.: 1 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES | ||||||||||||||||||||||||||||||||||||||||||
| Specimen support | Grid material: COPPER / Grid mesh size: 300 divisions/in. / Grid type: Quantifoil R1.2/1.3 | ||||||||||||||||||||||||||||||||||||||||||
| Vitrification | Instrument: LEICA EM GP / Cryogen name: ETHANE / Humidity: 90 % / Chamber temperature: 283 K |
- Electron microscopy imaging
Electron microscopy imaging
| Microscopy | Model: TFS TALOS |
|---|---|
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 36000 X / Nominal defocus max: 2200 nm / Nominal defocus min: 800 nm / Cs: 2.7 mm / C2 aperture diameter: 50 µm / Alignment procedure: COMA FREE |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Image recording | Average exposure time: 5 sec. / Electron dose: 53.112 e/Å2 / Film or detector model: GATAN K3 (6k x 4k) / Num. of grids imaged: 1 / Num. of real images: 3615 |
- Processing
Processing
| EM software | Name: PHENIX / Version: 1.21.1_5286 / Category: model refinement | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Image processing | Details: motioncorrected on the fly by cryosparc live | ||||||||||||||||||||||||
| CTF correction | Details: as implemented in cryosparc live / Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||
| Particle selection | Num. of particles selected: 2100293 / Details: topaz | ||||||||||||||||||||||||
| Symmetry | Point symmetry: C1 (asymmetric) | ||||||||||||||||||||||||
| 3D reconstruction | Resolution: 3.06 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 377557 / Algorithm: FOURIER SPACE / Details: as in cryosparc / Num. of class averages: 1 / Symmetry type: POINT | ||||||||||||||||||||||||
| Atomic model building | B value: 110.5 / Protocol: OTHER / Space: REAL / Target criteria: real space correlation | ||||||||||||||||||||||||
| Atomic model building | 3D fitting-ID: 1 / Accession code: 6V0O / Initial refinement model-ID: 1 / PDB-ID: 6V0O / Source name: PDB / Type: experimental model
| ||||||||||||||||||||||||
| Refinement | Cross valid method: NONE Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2 | ||||||||||||||||||||||||
| Displacement parameters | Biso mean: 114.39 Å2 | ||||||||||||||||||||||||
| Refine LS restraints |
|

