Journal: Nature / Year: 2025 Title: De novo design of transmembrane fluorescence-activating proteins. Authors: Jingyi Zhu / Mingfu Liang / Ke Sun / Yu Wei / Ruiying Guo / Lijing Zhang / Junhui Shi / Dan Ma / Qi Hu / Gaoxingyu Huang / Peilong Lu / Abstract: The recognition of ligands by transmembrane proteins is essential for the exchange of materials, energy and information across biological membranes. Progress has been made in the de novo design of ...The recognition of ligands by transmembrane proteins is essential for the exchange of materials, energy and information across biological membranes. Progress has been made in the de novo design of transmembrane proteins, as well as in designing water-soluble proteins to bind small molecules, but de novo design of transmembrane proteins that tightly and specifically bind to small molecules remains an outstanding challenge. Here we present the accurate design of ligand-binding transmembrane proteins by integrating deep learning and energy-based methods. We designed pre-organized ligand-binding pockets in high-quality four-helix backbones for a fluorogenic ligand, and generated a transmembrane span using gradient-guided hallucination. The designer transmembrane proteins specifically activated fluorescence of the target fluorophore with mid-nanomolar affinity, exhibiting higher brightness and quantum yield compared to those of enhanced green fluorescent protein. These proteins were highly active in the membrane fraction of live bacterial and eukaryotic cells following expression. The crystal and cryogenic electron microscopy structures of the designer protein-ligand complexes were very close to the structures of the design models. We showed that the interactions between ligands and transmembrane proteins within the membrane can be accurately designed. Our work paves the way for the creation of new functional transmembrane proteins, with a wide range of applications including imaging, ligand sensing and membrane transport.
Mass: 21460.938 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) artificial sequences (others) / Production host: Escherichia coli (E. coli)
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors China, 1items
China, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads


 PDBj
PDBj
 Assembly
Assembly



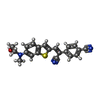
 Mass: 359.444 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H17N3OS / Feature type: SUBJECT OF INVESTIGATION
Mass: 359.444 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H17N3OS / Feature type: SUBJECT OF INVESTIGATION Mass: 18.015 Da / Num. of mol.: 31 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 31 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing