 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8v04: High resolution TMPRSS2 structure following acylation by nafamostat -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8v04 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | High resolution TMPRSS2 structure following acylation by nafamostat | |||||||||
 Components Components | (Transmembrane protease serine ...) x 2 | |||||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / Inhibitor complex / protease / viral entry / Structural Genomics / Structural Genomics Consortium / SGC / HYDROLASE / HYDROLASE-HYDROLASE INHIBITOR complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationtransmembrane protease serine 2 / protein autoprocessing / Attachment and Entry / serine-type peptidase activity / Induction of Cell-Cell Fusion / positive regulation of viral entry into host cell / Attachment and Entry / entry receptor-mediated virion attachment to host cell / serine-type endopeptidase activity / proteolysis ...transmembrane protease serine 2 / protein autoprocessing / Attachment and Entry / serine-type peptidase activity / Induction of Cell-Cell Fusion / positive regulation of viral entry into host cell / Attachment and Entry / entry receptor-mediated virion attachment to host cell / serine-type endopeptidase activity / proteolysis / extracellular exosome / extracellular region / plasma membrane Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.58 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.58 Å | |||||||||
 Authors Authors | Fraser, B.J. / Dong, A. / Kutera, M. / Seitova, A. / Li, Y. / Hutchinson, A. / Edwards, A. / Benard, F. / Levon, H. / Arrowsmith, C. | |||||||||
| Funding support |  Canada, Canada,  United States, 2items United States, 2items
| |||||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2025 Title: Large Library Docking and Biophysical Analysis of Small-Molecule TMPRSS2 Inhibitors. Authors: Fraser, B.J. / Young, N.J. / Bender, B.J. / Gahbauer, S. / Ilyassov, O. / Wilson, R.P. / Li, Y. / Seitova, A. / Lourenco, A.L. / Chung, D.H. / Bardine, C. / Benard, F. / Shoichet, B.K. / ...Authors: Fraser, B.J. / Young, N.J. / Bender, B.J. / Gahbauer, S. / Ilyassov, O. / Wilson, R.P. / Li, Y. / Seitova, A. / Lourenco, A.L. / Chung, D.H. / Bardine, C. / Benard, F. / Shoichet, B.K. / Craik, C.S. / Arrowsmith, C.H. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8v04.cif.gz | 102.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8v04.ent.gz | 72.4 KB | Display | PDB format |
| PDBx/mmJSON format | 8v04.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v0/8v04ftp://data.pdbj.org/pub/pdb/validation_reports/v0/8v04 | HTTPS FTP |
|---|
-Related structure data


-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Transmembrane protease serine ... , 2 types, 2 molecules AB
| #1: Protein | Mass: 12264.598 Da / Num. of mol.: 1 / Mutation: S251D, R252D, Q253D, S254D, R255K Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: TMPRSS2, PRSS10 / Plasmid: pFHMSP-LIC-CDetails (production host): baculovirus donor vector for secreted protein production Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: O15393 Spodoptera frugiperda (fall armyworm) / References: UniProt: O15393 |
|---|---|
| #2: Protein | Mass: 27856.742 Da / Num. of mol.: 1 / Fragment: Peptidase S1 domain residues 256-492 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: TMPRSS2 / Plasmid: pFHMSP-LIC-CDetails (production host): baculovirus donor vector for secreted protein production Production host: Spodoptera frugiperda (fall armyworm) / References: UniProt: O15393 |
-Sugars , 1 types, 1 molecules
| #3: Polysaccharide | 2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)- ...2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-[alpha-L-fucopyranose-(1-6)]2-acetamido-2-deoxy-beta-D-glucopyranose Type: oligosaccharide / Mass: 773.734 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source |
|---|
-Non-polymers , 7 types, 334 molecules 

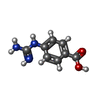




| #4: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 9 Mass: 62.068 Da / Num. of mol.: 9Source method: isolated from a genetically manipulated source Formula: C2H6O2 #5: Chemical | ChemComp-UNX / |  Mass: 179.176 Da / Num. of mol.: 1 / Source method: obtained synthetically Mass: 179.176 Da / Num. of mol.: 1 / Source method: obtained synthetically#6: Chemical | ChemComp-GBS / |  Mass: 179.176 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H9N3O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 179.176 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H9N3O2 / Feature type: SUBJECT OF INVESTIGATION#7: Chemical | ChemComp-CIT / |  Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7#8: Chemical | ChemComp-CA / |  Mass: 40.078 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: Ca#9: Chemical | ChemComp-PG4 / |  Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#10: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 320 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.52 Å3/Da / Density % sol: 51.28 % / Description: Hexagonal plates |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 4.6 Details: 3 uL hanging drop (2:1 protein:precipitant) grown over precipitant solution containing 25%PEG4000, 0.2M ammonium sulfate, and 0.1M sodium acetate pH 4.6. Protein (10 mg/mL) was in a buffer ...Details: 3 uL hanging drop (2:1 protein:precipitant) grown over precipitant solution containing 25%PEG4000, 0.2M ammonium sulfate, and 0.1M sodium acetate pH 4.6. Protein (10 mg/mL) was in a buffer containing 25 mM Tris pH 8.0, 75 mM NaCl, and 2 mM CaCl2 |
-Data collection
| Diffraction | Mean temperature: 100 K / Ambient temp details: cryo cooled / Serial crystal experiment: N | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 24-ID-C / Wavelength: 0.97918 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS EIGER2 X 16M / Detector: PIXEL / Date: Mar 19, 2022 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Cryo-cooled double crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97918 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.58→40 Å / Num. obs: 94364 / % possible obs: 92.8 % / Redundancy: 2.2 % / CC1/2: 0.988 / CC star: 0.997 / Rmerge(I) obs: 0.099 / Rpim(I) all: 0.079 / Rrim(I) all: 0.127 / Χ2: 2.246 / Net I/σ(I): 8.7 / Num. measured all: 212101 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.58→39.81 Å / Cor.coef. Fo:Fc: 0.972 / Cor.coef. Fo:Fc free: 0.96 / SU B: 1.615 / SU ML: 0.056 / Cross valid method: THROUGHOUT / ESU R: 0.075 / ESU R Free: 0.075 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.803 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.58→39.81 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|