 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8qj3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Receptor Sd-Amt1 (OFF-state) | |||||||||
 Components Components | Ammonium transporter | |||||||||
 Keywords Keywords | SIGNALING PROTEIN / Ammonium receptor / OFF-state / Shewanella denitrificans / Sd-Amt1 / diguanylate cyclase / Amt | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |  Shewanella denitrificans OS217 (bacteria) Shewanella denitrificans OS217 (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | |||||||||
 Authors Authors | Andrade, S.L. / Pflueger, T. / Gschell, M. | |||||||||
| Funding support |  Germany, 2items Germany, 2items
| |||||||||
 Citation Citation | Journal: Sci Adv / Year: 2024 Title: How sensor Amt-like proteins integrate ammonium signals. Authors: Tobias Pflüger / Mathias Gschell / Lin Zhang / Volodymyr Shnitsar / Annas J Zabadné / Paul Zierep / Stefan Günther / Oliver Einsle / Susana L A Andrade / Abstract: Unlike aquaporins or potassium channels, ammonium transporters (Amts) uniquely discriminate ammonium from potassium and water. This feature has certainly contributed to their repurposing as ammonium ...Unlike aquaporins or potassium channels, ammonium transporters (Amts) uniquely discriminate ammonium from potassium and water. This feature has certainly contributed to their repurposing as ammonium receptors during evolution. Here, we describe the ammonium receptor Sd-Amt1, where an Amt module connects to a cytoplasmic diguanylate cyclase transducer module via an HAMP domain. Structures of the protein with and without bound ammonium were determined to 1.7- and 1.9-Ångstrom resolution, depicting the ON and OFF states of the receptor and confirming the presence of a binding site for two ammonium cations that is pivotal for signal perception and receptor activation. The transducer domain was disordered in the crystals, and an AlphaFold2 prediction suggests that the helices linking both domains are flexible. While the sensor domain retains the trimeric fold formed by all Amt family members, the HAMP domains interact as pairs and serve to dimerize the transducer domain upon activation. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8qj3.cif.gz | 333 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8qj3.ent.gz | 266.4 KB | Display | PDB format |
| PDBx/mmJSON format | 8qj3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qj/8qj3ftp://data.pdbj.org/pub/pdb/validation_reports/qj/8qj3 | HTTPS FTP |
|---|
-Related structure data



-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| 2 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 70836.547 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Shewanella denitrificans OS217 (bacteria)Strain: OS217 / Gene: Sden_0766 / Details (production host): C-terminal His-tag / Production host: #2: Chemical |   Mass: 35.453 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: Cl#3: Sugar | 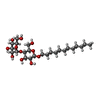  Type: D-saccharide / Mass: 510.615 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H46O11 / Comment: detergent*YM Type: D-saccharide / Mass: 510.615 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H46O11 / Comment: detergent*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 373 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 373 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.49 Å3/Da / Density % sol: 50.63 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 6.5 / Details: 0.1 M Bis/Tris pH 6.5 with 25 % (w/v) PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 0.976 Å / Beamline: X06SA / Wavelength: 0.976 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Dec 25, 2021 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.976 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→93.61 Å / Num. obs: 107695 / % possible obs: 99.8 % / Redundancy: 5.8 % / CC1/2: 0.999 / Rmerge(I) obs: 0.072 / Rpim(I) all: 0.033 / Rrim(I) all: 0.08 / Net I/σ(I): 14.8 / Num. measured all: 624924 |
| Reflection shell | Resolution: 1.9→1.93 Å / % possible obs: 97.4 % / Redundancy: 5.7 % / Rmerge(I) obs: 1.351 / Num. measured all: 29533 / Num. unique obs: 5208 / CC1/2: 0.404 / Rpim(I) all: 0.621 / Rrim(I) all: 1.487 / Net I/σ(I) obs: 1.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.9→48.97 Å / Cor.coef. Fo:Fc: 0.957 / Cor.coef. Fo:Fc free: 0.943 / SU B: 4.739 / SU ML: 0.065 / Cross valid method: THROUGHOUT / ESU R: 0.094 / ESU R Free: 0.092 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.026 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.9→48.97 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|