 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8q77: STRUCTURE OF PROTEIN KINASE CK2 CATALYTIC SUBUNIT (ISOFORM CK2ALP... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8q77 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF PROTEIN KINASE CK2 CATALYTIC SUBUNIT (ISOFORM CK2ALPHA'; CSNK2A2 GENE PRODUCT) IN COMPLEX WITH THE BISUBSTRATE INHIBITOR ARC-780 | |||||||||
 Components Components | Casein kinase II subunit alpha' | |||||||||
 Keywords Keywords | TRANSFERASE / protein kinase CK2 casein kinase 2 bisubstrate inhibitor N-terminal segment ligand binding site | |||||||||
| Function / homology |  Function and homology information Function and homology informationregulation of mitophagy / regulation of chromosome separation / Condensation of Prometaphase Chromosomes / WNT mediated activation of DVL / protein kinase CK2 complex / positive regulation of protein targeting to mitochondrion / Receptor Mediated Mitophagy / Synthesis of PC / Maturation of hRSV A proteins / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known ...regulation of mitophagy / regulation of chromosome separation / Condensation of Prometaphase Chromosomes / WNT mediated activation of DVL / protein kinase CK2 complex / positive regulation of protein targeting to mitochondrion / Receptor Mediated Mitophagy / Synthesis of PC / Maturation of hRSV A proteins / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known / negative regulation of apoptotic signaling pathway / negative regulation of proteasomal ubiquitin-dependent protein catabolic process / liver regeneration / acrosomal vesicle / Signal transduction by L1 / Regulation of PTEN stability and activity / cerebral cortex development / Wnt signaling pathway / KEAP1-NFE2L2 pathway / Cooperation of PDCL (PhLP1) and TRiC/CCT in G-protein beta folding / double-strand break repair / Regulation of TP53 Activity through Phosphorylation / spermatogenesis / non-specific serine/threonine protein kinase / protein serine kinase activity / protein serine/threonine kinase activity / apoptotic process / DNA damage response / chromatin / nucleoplasm / ATP binding / nucleus / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.255 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.255 Å | |||||||||
 Authors Authors | Werner, C. / Lindenblatt, D. / Niefind, K. | |||||||||
| Funding support |  Germany, 2items Germany, 2items
| |||||||||
 Citation Citation | Journal: Kinases Phosphatases / Year: 2023 Title: Discovery and Exploration of Protein Kinase CK2 Binding Sites Using CK2alpha Cys336Ser as an Exquisite Crystallographic Tool Authors: Werner, C. / Lindenblatt, D. / Viht, K. / Uri, A. / Niefind, K. #1: Journal: ACS Omega / Year: 2019Title: Diacritic Binding of an Indenoindole Inhibitor by CK2alpha Paralogs Explored by a Reliable Path to Atomic Resolution CK2alpha' Structures. Authors: Lindenblatt, D. / Nickelsen, A. / Applegate, V.M. / Hochscherf, J. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8q77.cif.gz | 284.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8q77.ent.gz | 190.5 KB | Display | PDB format |
| PDBx/mmJSON format | 8q77.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 8q77_validation.pdf.gz | 902.2 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 8q77_full_validation.pdf.gz | 911.2 KB | Display | |
| Data in XML | 8q77_validation.xml.gz | 19.1 KB | Display | |
| Data in CIF | 8q77_validation.cif.gz | 28.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q7/8q77ftp://data.pdbj.org/pub/pdb/validation_reports/q7/8q77 | HTTPS FTP |
-Related structure data





-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 42879.867 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CSNK2A2, CK2A2 / Production host:  References: UniProt: P19784, non-specific serine/threonine protein kinase | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | 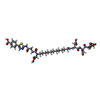  Mass: 920.936 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C40H52N6O17S / Feature type: SUBJECT OF INVESTIGATION Mass: 920.936 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C40H52N6O17S / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-EDO /   Mass: 62.068 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical | ChemComp-CL / |   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 311 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 311 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.66 Å3/Da / Density % sol: 53.84 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: THE CK2ALPHA' SOLUTION AFTER PROTEIN PURIFICATION (5 MG/ML CK2ALPHA' IN 500 MM NACL, 25 MM TRIS/HCl, PH 8.5) WAS MIXED IN 1:10 VOLUME RATIO WITH 10 MM SOLUTION OF THE INHIBITOR MB002 IN DMSO. ...Details: THE CK2ALPHA' SOLUTION AFTER PROTEIN PURIFICATION (5 MG/ML CK2ALPHA' IN 500 MM NACL, 25 MM TRIS/HCl, PH 8.5) WAS MIXED IN 1:10 VOLUME RATIO WITH 10 MM SOLUTION OF THE INHIBITOR MB002 IN DMSO. THIS SOLUTION WAS MIXED IN 2:1 RATIO WITH RESERVOIR SOLUTION [900 MM LICL, 28 % (W/V) PEG 6000, 250 MM TRIS/HCL, PH 8.5] IN SITTING DROP PLATES (VAPOUR DIFFUSION). THE CRYSTAL GROWTH WAS INDUCED BY MICROSEEDING. THE CRYSTALS WERE OPTIMIZED BY MACROSEEDING AND PURGED TWICE WITH RESERVOIR SOLUTION. THEN, ARC-780 WAS ADDED TO AN INITIAL CONCENTRATION OF 1 MM BY ADDING 1 MIKROLITER OF A 10 MM ARC-780 SOLUTION IN DMSO IN A 1:10 RATIO TO THE DROPLET. AFTER EXTENSIVE INTIAL SOAKING, THE SALT CONCENTRATION WAS SUCCESSIVELY LOWERED OVER A PERIOD OF 8 MONTHS WHILE IN PARALLEL THE CONCENTRATIONS OF DMSO AND PEG 6000 WERE INCREASED. THIS WAS DONE BY GRADUALLY REPLACING THE RESERVOIR AND THE DROPLET SOLUTION UNTIL ANY NACL WAS REMOVED, THE LICL CONCENTRATION WAS REDUCED TO 50 MM, THE PEG 6000 CONCENTRATION WAS INCREASED TO SATURATION AND A DMSO CONTENT OF 30% WAS REACHED. MEANWHILE, THE ARC-780 LIGAND WAS INCLUDED IN ANY DESALTING STEP REACHING SATURATION AFTER THE FINAL DILUTION STEP. ALL STEPS WERE PERFORMED AT A TEMPERATURE OF 293 K. |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: MASSIF-3 / Wavelength: 0.9677 Å / Beamline: MASSIF-3 / Wavelength: 0.9677 Å |
| Detector | Type: DECTRIS EIGER X 4M / Detector: PIXEL / Date: Nov 14, 2020 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9677 Å / Relative weight: 1 |
| Reflection | Resolution: 1.255→46.578 Å / Num. obs: 80300 / % possible obs: 72.5 % / Redundancy: 3.9 % / Biso Wilson estimate: 10.32 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.052 / Rpim(I) all: 0.031 / Rrim(I) all: 0.061 / Rsym value: 0.052 / Net I/σ(I): 12.2 |
| Reflection shell | Resolution: 1.255→1.364 Å / Rmerge(I) obs: 0.559 / Mean I/σ(I) obs: 1.6 / Num. unique obs: 4015 / CC1/2: 0.806 / Rpim(I) all: 0.326 / Rrim(I) all: 0.648 / Rsym value: 0.559 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.255→41.25 Å / SU ML: 0.0864 / Cross valid method: FREE R-VALUE / σ(F): 2 / Phase error: 16.9147 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.47 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.255→41.25 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
