 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8q2b: E. coli Adenylate Kinase variant D158A (AK D158A) showing signifi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8q2b | ||||||
|---|---|---|---|---|---|---|---|
| Title | E. coli Adenylate Kinase variant D158A (AK D158A) showing significant changes to the stacking of catalytic arginine side chains | ||||||
 Components Components | Adenylate kinase | ||||||
 Keywords Keywords | TRANSFERASE / PHOSPHOTRANSFERASE / ADENYLATE KINASE / D158A VARIANT / AP5A LIGAND / PROTEIN DYNAMICS | ||||||
| Function / homology |  Function and homology information Function and homology informationadenylate kinase / AMP kinase activity / AMP salvage / ATP binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.76 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.76 Å | ||||||
 Authors Authors | Sauer, U.H. / Wolf-Watz, M. / Nam, K. | ||||||
| Funding support |  Sweden, 1items Sweden, 1items
| ||||||
 Citation Citation | Journal: J.Chem.Inf.Model. / Year: 2024 Title: Elucidating Dynamics of Adenylate Kinase from Enzyme Opening to Ligand Release. Authors: Nam, K. / Arattu Thodika, A.R. / Grundstrom, C. / Sauer, U.H. / Wolf-Watz, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8q2b.cif.gz | 183.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8q2b.ent.gz | 146.8 KB | Display | PDB format |
| PDBx/mmJSON format | 8q2b.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q2/8q2bftp://data.pdbj.org/pub/pdb/validation_reports/q2/8q2b | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data | |
| Experimental dataset #1 | Data reference: 10.1016/s0969-2126(96)00018-4 / Data set type: other data |

-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23576.020 Da / Num. of mol.: 2 / Mutation: D158A Source method: isolated from a genetically manipulated source Details: AK D158A variant / Source: (gene. exp.) #2: Chemical | 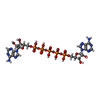  Mass: 916.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H29N10O22P5 / Feature type: SUBJECT OF INVESTIGATION Mass: 916.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H29N10O22P5 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-SO4 / |   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-MPO / |   Mass: 209.263 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H15NO4S / Comment: pH buffer*YM Mass: 209.263 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H15NO4S / Comment: pH buffer*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 456 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 456 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.84 Å3/Da / Density % sol: 56.62 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: Crystallization: 2 ul of purified AK-D158A at 16.4 mg/ml, mixed with the non-hydrolysable inhibitor Ap5A at a final concentration of 5 mM and 2 ul of precipitant buffer containing 32% PEG ...Details: Crystallization: 2 ul of purified AK-D158A at 16.4 mg/ml, mixed with the non-hydrolysable inhibitor Ap5A at a final concentration of 5 mM and 2 ul of precipitant buffer containing 32% PEG 8K, 0.2 M AmSO4, 0.1 M Cacodylate at pH 6.5. |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 0.87293 Å / Beamline: ID23-2 / Wavelength: 0.87293 Å |
| Detector | Type: DECTRIS PILATUS3 X 6M / Detector: PIXEL / Date: May 2, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.87293 Å / Relative weight: 1 |
| Reflection | Resolution: 1.76→47.45 Å / Num. obs: 52176 / % possible obs: 100 % / Redundancy: 6.9 % / Biso Wilson estimate: 22.1 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.109 / Rpim(I) all: 0.045 / Rrim(I) all: 0.118 / Net I/σ(I): 11.2 |
| Reflection shell | Resolution: 1.76→1.82 Å / Redundancy: 7.1 % / Rmerge(I) obs: 1.097 / Mean I/σ(I) obs: 1.82 / Num. unique obs: 5114 / CC1/2: 0.627 / Rpim(I) all: 0.44 / Rrim(I) all: 1.183 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.76→40.08 Å / SU ML: 0.19 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 22.02 / Stereochemistry target values: ML Details: Iterative rounds of AK-D158A structure refinement against data extending to 0.176 nm using phenix.refine (Afonine, 2010; Afonine, 2012) of the Phenix program package (version 1.19.2-4158-000) ...Details: Iterative rounds of AK-D158A structure refinement against data extending to 0.176 nm using phenix.refine (Afonine, 2010; Afonine, 2012) of the Phenix program package (version 1.19.2-4158-000) and manual model building with Coot (version 0.9.8.1-EL (Emsley, 2010)) were carried out until the R-free factor and R-factor converged.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.76→40.08 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
